the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 May 2018
| 03 May 2018
A steady-state continuous flow chamber for the study of daytime and nighttime chemistry under atmospherically relevant NO levels
John Ortega
Yuanlong Huang
Stephen Shertz
Geoffrey S. Tyndall
John J. Orlando
Experiments performed in laboratory chambers have contributed significantly to the understanding of the fundamental kinetics and mechanisms of the chemical reactions occurring in the atmosphere. Two chemical regimes, classified as “high-NO” vs. “zero-NO” conditions, have been extensively studied in previous chamber experiments. Results derived from these two chemical scenarios are widely parameterized in chemical transport models to represent key atmospheric processes in urban and pristine environments. As the anthropogenic NOx emissions in the United States have decreased remarkably in the past few decades, the classic “high-NO” and “zero-NO” conditions are no longer applicable to many regions that are constantly impacted by both polluted and background air masses. We present here the development and characterization of the NCAR Atmospheric Simulation Chamber, which is operated in steady-state continuous flow mode for the study of atmospheric chemistry under “intermediate NO” conditions. This particular chemical regime is characterized by constant sub-ppb levels of NO and can be created in the chamber by precise control of the inflow NO concentration and the ratio of chamber mixing to residence timescales. Over the range of conditions achievable in the chamber, the lifetime of peroxy radicals (RO2), a key intermediate from the atmospheric degradation of volatile organic compounds (VOCs), can be extended to several minutes, and a diverse array of reaction pathways, including unimolecular pathways and bimolecular reactions with NO and HO2, can thus be explored. Characterization experiments under photolytic and dark conditions were performed and, in conjunction with model predictions, provide a basis for interpretation of prevailing atmospheric processes in environments with intertwined biogenic and anthropogenic activities. We demonstrate the proof of concept of the steady-state continuous flow chamber operation through measurements of major first-generation products, methacrolein (MACR) and methyl vinyl ketone (MVK), from OH- and NO3-initiated oxidation of isoprene.
- Article
(2523 KB) -
Supplement
(3270 KB) - BibTeX
- EndNote





With the discovery of the role of biogenic volatile organic compounds (BVOCs) in urban photochemical smog (Chameides et al., 1988), the interactions of biogenic emissions with manmade pollution and their subsequent impact on the atmosphere's oxidative capacity and aerosol burden have received extensive studies in the ensuing decades (De Gouw et al., 2005; Ng et al., 2007; Goldstein et al., 2009; Surratt et al., 2010; Rollins et al., 2012; Shilling et al., 2013; Xu et al., 2015). A particular research focus has been understanding the influence of nitrogen oxides () on the atmospheric oxidation cascades of BVOCs, which ultimately generate ozone (O3) and secondary organic aerosols (SOA). Nitrogen oxides alter the distribution of BVOC oxidation products by primarily modulating the fate of peroxy radicals (RO2), a key intermediate produced from the atmospheric degradation of VOCs by major oxidants including OH, O3, and NO3. In the absence of NOx, RO2 reacts predominantly with HO2 radicals yielding organic peroxides and other products and, to a lesser extent, undergoes self- or cross-reactions yielding carbonyls, alcohols, and multifunctional species. In the presence of elevated NOx, the dominant fate of RO2 is to react with NO leading to ozone production, and also to organic nitrates. During the night, RO2 also reacts with NO3 which is produced by the reaction between O3 and NO2. In addition, reaction of peroxyacyl radicals (RC(O)O2) with NO2 produces peroxyacyl nitrates that constitute a large reservoir of reactive nitrogen and a potentially important SOA precursor (Singh and Hanst, 1981; Nguyen et al., 2015).
Much of our understanding of the extent to which NOx mediates the oxidation chemistry of BVOC in the atmosphere has been derived from measurements in laboratory chambers, where two extreme experimental conditions, i.e., “high-NO” vs. “zero-NO”, were mostly performed to examine the reaction pathways of RO2 radicals (Kroll and Seinfeld, 2008; Orlando and Tyndall, 2012; Ziemann and Atkinson, 2012). Results from these two chemical regimes have been widely incorporated into chemical transport models to represent key atmospheric processes in urban and pristine environments, respectively (Kanakidou et al., 2005). In the actual atmosphere, however, the fate of RO2 radicals is rather more complicated than simply undergoing bimolecular reactions with NO ∕ HO2 as observed under the two extreme chamber conditions. It has been recently revealed that RO2 radicals may undergo an internal H-shift followed by sequential O2 addition, leading to highly oxygenated multifunctional peroxides (Ehn et al., 2014; Jokinen et al., 2015; Kurtén et al., 2015; Kirkby et al., 2016; Zhang et al., 2017). The rate of H-shift largely depends on the thermochemistry of the nascent alkyl radicals and can be reasonably fast, on a timescale of seconds to minutes (Crounse et al., 2013). Further, depending on the stability of the RO2 precursor (alkyl radicals), RO2 radicals may lose O2 in competition with bimolecular reactions with NO, NO3, RO2, and HO2. Recent theoretical and laboratory studies have found that the hydroxy peroxy radical conformers produced from isoprene photooxidation decompose readily to allylic radicals on timescales faster than bimolecular processes under atmospherically relevant NO and HO2 levels (tens to hundreds of parts per trillion by volume). This highly dynamic system leads to formation of distinctly different products that depend on the concentrations of bimolecular reaction partners from those observed in chamber experiments under “high-NO” and “zero-NO” conditions (Teng et al., 2017).
Anthropogenic NOx emissions in the United States have decreased remarkably in the past few decades (EPA, 2014), resulting in significant changes in the degradation mechanisms of BVOCs, especially in regions impacted by both background and polluted air masses such as the southeastern United States. However, the ultimate fate of peroxy radicals in environments with sub-ppb NO levels is still poorly constrained, in part due to a lack of consistent measurements under well-controlled conditions. Experimental approaches targeting a controlled NO level (sub-ppb to ppb) have been introduced over the years. For outdoor chambers, experiments were typically performed by exposing a gas mixture of O3 ∕ NOx ∕ VOCs or HONO ∕ NOx ∕ VOCs to natural sunlight (Bloss et al., 2005; Karl et al., 2006). OH radicals were produced either via the photolysis of ozone and subsequent reaction of O(1D) with H2O or directly from the photolysis of HONO. NO levels ranging from a few hundreds of ppt to a few ppb over the course of several hours of reactions have been reported. In the absence of any additional supply, NO will be eventually depleted in a closed chamber environment, and the initial “moderate-NO” condition will essentially transfer to the “zero-NO” condition. For indoor chambers, a “slow chemistry” scenario initiated by photolyzing methyl nitrite (CH3ONO) under extremely low UV intensities as the OH radical source ( s−1) was created to study the autoxidation chemistry of peroxy radicals produced from isoprene photooxidation (Crounse et al., 2011, 2012; Teng et al., 2017). The resulting NO and HO2 mixing ratios are maintained at ∼ ppt level () over the course of several hours of reaction, and the average OH concentration (OH∼105 molec cm−3) is approximately 1 order of magnitude lower than that in the typical daytime ambient atmosphere. Another example relates to a recent method development in the potential aerosol mass (PAM) flow tube reactor where nitrous oxide (N2O) was used to produce ∼ ppb level of NO (; ) (Lambe et al., 2017). Timescales for chemical reactions and gas–particle partitioning are ultimately limited to the mean residence time (∼80 s) of the PAM reactor.
An alternative experimental platform to the batch-mode chamber and flow tube reactor described above is a well-mixed steady-state chamber with continuous feed of reactants and continuous withdrawal of reactor contents (Shilling et al., 2008). An attribute of the continuous flow steady-state chamber is that, by control of the inlet reactant concentrations and the ratio of mixing to residence timescales, it is possible to simulate atmospheric oxidation under stable conditions over a wide range of time periods and chemical scenarios. For example, a steady-state NO level at ∼1 ppb was created by the continuously mixed flow chamber operation for the study of isoprene photooxidation chemistry (Liu et al., 2013). In this study, we present the development and characterization of the NCAR Atmospheric Simulation Chamber, which is operated in steady-state continuous flow mode for simulating atmospheric daytime and nighttime chemistry over chemical regimes not accessible in static chamber experiments. We focus on establishing an “intermediate NO” regime characterized by a constant steady-state NO level ranging from tens of ppt to a few ppb in the chamber. This particular chemical regime is well suited for the study of atmospheric behavior of RO2 radicals, as they can survive up to minutes and embrace various reaction possibilities as opposed to reaction with NO, NO3, HO2, and RO2 as their dominant fate observed in most batch-mode chamber experiments. We employ the “intermediate NO” regimes to reexamine the daytime and nocturnal chemistry of isoprene through the measurements of two first-generation products, methacrolein (MACR) and methyl vinyl ketone (MVK).
2.1 NCAR Atmospheric Simulation Chamber facilities
The NCAR Atmospheric Simulation Chamber consists of a 10 m3 FEP Teflon (0.005 in. thick) bag that is housed in a cubic enclosure with UV reflective surfaces and a bank of 128 wall-mounted blacklight tubes (32 W, type F32T8/BL). To characterize photolytic conditions in the chamber, irradiance spectra were collected in the wavelength range of 180–600 nm at ∼0.8 nm resolution by a custom-built spectroradiometer, as shown in Fig. S1 in the Supplement (Petropavlovskikh et al., 2007). Photolysis frequencies were calculated based on the measured downwelling spectral actinic fluxes. The computed photolysis rate of NO2 () agrees within 3 % with that measured by photolyzing 18.6 ppb NO2 in the chamber and monitoring the NO production rate. The chamber is equipped with a standard set of measurements, including an integrated temperature and humidity probe (model 50U, VAISALA, CO) and a Magnehelic differential pressure indicating transmitter (model 605-11, Dwyer Instruments, IN). The chamber temperature is controlled at 295 K by the building's air conditioning system and increases to 305–306 K under maximum irradiation conditions. The relative humidity of the chamber air is below 10 % under dry conditions (the remaining water vapor is generated from methane combustion during the air purification process) and can be varied in the range of ∼10–50 % by flowing a portion of the purified dry flushing air into the chamber through a temperature-controlled water reservoir. Typical temperature and relative humidity profiles across the chamber under maximum irradiation conditions are given in Fig. S1 in the Supplement. The chamber internal pressure is maintained slightly above the ambient pressure to minimize the enclosure air contamination via penetration through the Teflon film.
Prior to each experiment, the chamber was flushed with purified dry air from an ultra high purity zero air generator (model 737, Aadco Instruments, OH) for >12 h until ozone and NOx levels were below 1 ppb. During the operation of the steady-state continuous flow mode, the chamber was constantly flushed with purified dry air at 40 L min−1, which gives an average chamber residence time of 4.17 h. The incoming and outgoing flows were balanced by a feedback control system that maintains a constant internal pressure of 1.2– atm above the ambient. The chamber is actively mixed by the turbulence created by the 40 L min−1 flushing air. The characteristic mixing time is defined as the time it takes for the measurement signal of a tracer compound (e.g., CO2 and NO) to stabilize following a pulse injection. The average mixing time in the NCAR chamber was determined to be ∼9 min, which is ∼4 % of the residence time. Under such conditions, the gas- and particle-phase composition in the outflow can be assumed identical to that in the well-mixed core of the chamber.
To mimic daytime photochemistry in the continuous flow mode, steady-state OH mixing ratio was created by photolyzing hydrogen peroxide (H2O2) vapor that was continuously flowing into the chamber (, ). Specifically, a 20 mL syringe (NORM-JECT, Henke-Sass Wolf, MA) held on a syringe pump (model 100, KD Scientific, MA) kept at ∼4 ∘C was used to deliver H2O2 solution (1–30 wt %, Sigma Aldrich, MO) into a glass bulb that was gently warmed at ∼32 ∘C. The liquid delivery rate was sufficiently slow (100–300 µL min−1) that all the H2O2 vapor was released into the glass bulb through evaporation of a small droplet hanging on the needle tip. An air stream (5 L min−1) swept the H2O2 vapor into the chamber, resulting in an H2O2 mixing ratio in the range of 600 ppb to 16.22 ppm in the injection flow as a function of the concentration of H2O2 aqueous solution used. A spreadsheet (Table S2) for calculating the inflow H2O2 mixing ratio using the above input method is provided in the Supplement. As H2O2-laden air was continuously entering the chamber, it took approximately three turnover times (∼12.5 h) for the desired H2O2 vapor mixing ratio to reach steady state in the chamber. The H2O2 vapor concentration in the chamber, though not measured, can be estimated from the steady-state OH mixing ratio derived from the observed exponential decay of a given parent hydrocarbon. Constant NO injection flow was achieved by diluting the gas flow from a concentrated NO cylinder (NO =133.16 ppm, balance N2) to a desired mixing ratio (0.1–100 ppb) using a set of mass flow controllers (Tylan FC260 and FC262, Mykrolis Corp., MA). The lowest steady-state NO level that can be achieved in the chamber is around 30 ppt (unpublished, NCAR). Note that for experiments performed in the absence of any VOC precursor, H2O2 and NO were the only two species that were continuously input into the chamber for the establishment of a combination of different photochemical conditions as denoted by the O3 and NOx measurements. For the isoprene photooxidation experiments, an isoprene standard (C5H8=531 ppm, balance N2) was constantly injected into the chamber and diluted with the flushing air to achieve an inflow concentration of ∼20 ppb.
To mimic the nighttime chemistry in the continuous flow mode, steady-state NO3 mixing ratio was created by constantly flowing diluted O3 and NO air into the chamber (; ). The NO source can be replaced by NO2, although the absolute absence of NO does not necessarily represent the actual atmospheric conditions. O3 was produced by photolyzing O2 in air at 185 nm using a mercury Pen-Ray lamp (UVP LLC, CA). Ozone concentration in the injection flow can be controlled from 3.5 to 457 ppb automatically by adjusting the mercury lamp duty cycle. To study the RO2+HO2 pathway, formaldehyde (CH2O) was input into the chamber along with NO and O3 to initiate HO2 production (). Formaldehyde aqueous solution (37 wt %, Sigma Aldrich, MO) was diluted with ultrapure water (Milli-Q, Merck Millipore, MA) to 0.2–1.0 wt % and continuously input into the chamber using the same method used for H2O2 input described above. It is worth noting that the formaldehyde aqueous solution contains 10–15 % methanol as stabilizer to prevent polymerization. The presence of methanol in the chamber does not significantly impact the nocturnal chemistry as it consumes OH and NO3 radicals to generate formaldehyde and additional HO2 (, ) (Atkinson et al., 2006). The use of formaldehyde as an HO2 source mimics closely the atmospheric nighttime conditions in forest environments (Schwantes et al., 2015). To study the NO3-initiated oxidation of isoprene, an injection flow of diluted isoprene (∼ 10 ppb) was achieved using the procedure described above.
2.2 Analytical measurements
A suite of instruments was used to monitor gas-phase concentrations in the chamber outflow. O3 was monitored by absorption spectroscopy with 0.5 ppb detection limit (model 49, Thermo Scientific, CO). The O3 monitor was calibrated using an Ozone Primary Standard in the range of 0–200 ppb (model 49i-PS, Thermo Scientific, CO). The O3 monitor used for chamber experiments was periodically checked with the primary standard and was shown to be stable over long periods of time (less than 1 ppb drift in over 1 year). NO was monitored by chemiluminescence with 0.5 ppb detection limit (model CLD 88Y, Eco Physics, MI). Zero-point and span calibrations of the NOx monitor were performed prior to each experiment by supplying the instrument with pure N2 gas and diluted NO, respectively. Multi-point calibration was performed on a weekly basis and has shown a good stability and linearity in the NO measurement ranging from 1 to 200 ppb. NOx (NO+NO2) measurements were performed using a photolytic converter that selectively converts NO2 to NO upstream of the photo-multiplier tube in the CLD 88Y NO monitor. This converter uses two opposing arrays of UV LEDs shining into a cylindrical quartz mixing tube to achieve approximately 50 % conversion of NO2 to NO per second. The total efficiency for the equipment described here is approximately 70 % as determined by measuring calibrated NO2 standards. The sample path always includes the photolytic converter, and typical experiments cycle the power for the LED lights to switch between measuring NO (lights off) and NOx (lights on). NO2 concentrations were then determined by subtracting the NO from the adjacent NOx measurements.
A customized proton transfer reaction quadrupole mass spectrometer (PTR-Q-MS) was used to measure volatile organic compounds including isoprene (C5H8), MACR (C4H6O), and MVK (C4H6O). The instrument was operated at 2.3 mbar drift pressure and 560 V drift voltage. Measurements reported here were obtained at a sampling rate of 10 Hz. In positive-mode operation, a given analyte [M] undergoes proton transfer reaction, producing an ion of the form [M+H]+; that is, isoprene is detected as ion (m∕z 69) and MACR and MVK are both detected as ion C4H7O+ (m∕z 71). The instrument background was collected by sampling the chamber air for at least 30 min prior to each experiment. Measured ion intensities for isoprene (, m∕z 69) and MACR and MVK (C4H7O+, m∕z 71) were calculated as the signal of each ion (counts per seconds) normalized to the total ion signal of H3O+. The instrument sensitivities towards isoprene, MACR, and MVK were calibrated with a mixture of diluted gas standards. The instrument sensitivity towards MACR is identical to that of MVK and, as a result, the sum of MACR and MVK concentration in the sampling air can be calculated by applying one calibration factor to the measured C4H7O+ (m∕z 71) signal intensity. Since artifacts in the measured C4H7O+ signal can be produced through thermal decomposition of isoprene oxidation products, such as the peroxides, nitrates, and epoxides, on contact with hot metal surface (Liu et al., 2013; Nguyen et al., 2014b; Rivera-Rios et al., 2014), a cold-trap system was used to avoid bias in the interpretation of the PTR-Q-MS data. Specifically, a 1 m section of Teflon tubing was submerged in a low-temperature ethanol bath (−40 ± 2 ∘C) that could trap oxidized products less volatile than the authentic MACR and MVK standards after steady state was established in the chamber. The quantification of the sum of MACR and MVK was then based on the PTR-Q-MS measured C4H7O+ (m∕z 71) signal downstream of the cold trap.
Reaction kinetics and mechanisms for the gas-phase photochemistry were extracted from the Master Chemical Mechanism (MCMv3.3.1, accessible at http://mcm.leeds.ac.uk/MCM/, last access: 4 January 2018). The inorganic reaction scheme includes 21 species and 48 reactions; the isoprene oxidation system includes 611 species and 1974 reactions. The kinetic schemes were implemented in MATLAB (Mathworks) to simulate the temporal profile of a given compound i in the chamber operated in the steady-state continuous flow mode:
where Ci (molec cm−3) is the gas-phase concentration of compound i in the well-mixed core of the chamber; Ci,0 (molec cm−3) is the initial gas-phase concentration of compound i in the injection flow; ki (s−1) is the pseudo-first-order rate coefficient for a chemical reaction that consumes compound i; τ (s) is the chamber mean residence time and can be calculated as the total chamber volume divided by the incoming and outgoing flow rate; and Pi (molec cm−3) is the increment in the concentration of compound i through chemical production during one residence time. Note that two terms are neglected in Eq. (1), i.e., organic vapor condensation onto particles and deposition on the chamber wall. This is a reasonable simplification here due to the relatively high volatility ( atm) of compounds studied (Zhang et al., 2015b; Krechmer et al., 2016; Huang et al., 2018). Incorporation of these two terms into Eq. (1) is feasible given the vapor pressure of compound i, suspended particle size distribution, gas–particle and gas–wall partitioning coefficient, accommodation coefficients of compound i on particles and walls, and the effective absorbing organic masses on the wall (Zhang et al., 2014a, 2015b; Huang et al., 2016; McVay et al., 2016; Nah et al., 2016).
Model simulations used for comparison with chamber measurements were initialized using experimental conditions summarized in Table S1 in the Supplement. Model input parameters for all simulations include temperature (295 K at dark and 306 K under irradiation), local pressure (8.6×104 Pa), relative humidity (8 % at dark and 5 % under irradiation), light intensity ( under irradiation and 0 at dark), chamber mean residence time (4.17 h), and input mixing ratios of H2O2 (0.11–16.2 ppm for photolytic experiments), NO (0.1–100 ppb for photolytic experiments and 10–20 ppb for dark experiments), O3 (22–225 ppb for dark experiments), HCHO (0–600 ppb for dark experiments), and isoprene (19.9 ppb for photolytic experiments and 10.2 ppb for dark experiments). The model was propagated numerically for 25 h duration for each experiment.
4.1 Optimal operating conditions for daytime photochemistry
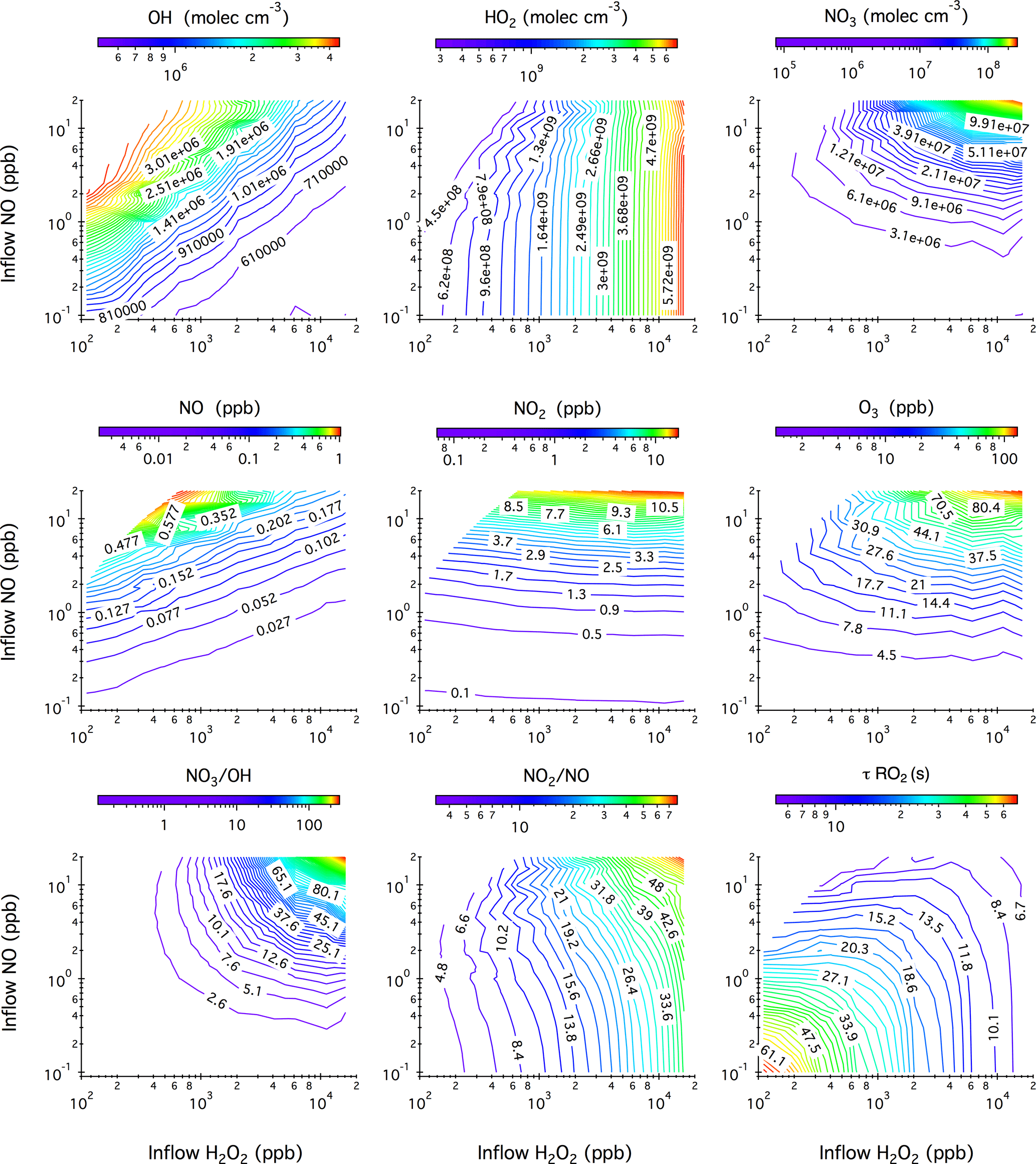
Figure 1Contour plots showing the model-predicted steady-state mixing ratios of OH, HO2, NO3, NO, NO2, and O3 after 20 h of photochemical reactions in the chamber as a function of the concentrations of H2O2 and NO in the continuous injection flow. Also given here are the simulated NO3 to OH ratio, NO2 to NO ratio, and the lifetime of an RO2 radical () with respect to reactions with NO and HO2. Note that the ripples on the contour lines originate from the limited simulation datasets that are used to generate iso-response values.
Figure 1 shows the model-predicted steady-state mixing ratios of OH, HO2, NO3, NO, NO2, and O3 in the chamber after 20 h of photochemical reactions as a function of the H2O2 and NO concentrations in the continuous injection flow. Six blank chamber experiments were compared with simulations. In general, the model captures the evolution patterns of NOx and O3 well. The predicted mixing ratios of NO, NO2, and O3 agree within 69, 11, and 33 %, respectively, with the measurements (see Table S1 and Fig. S2 in the Supplement). The relatively large NO uncertainties originate from the measurements that were performed close to the instrument detection limit (0.5 ppb).
Simulated steady-state mixing ratios of OH radicals ([OH]ss) range from to molec cm−3, which over ∼4 h chamber residence time would be roughly equivalent to ∼1 to ∼8 h of atmospheric OH exposure (1×106 molec cm−3). As expected, [OH]ss increases with increasing NO influxes due to the enhanced NOx ∕ O3 cycling but decreases with increasing H2O2 influxes due to the overwhelming reaction . As a consequence, the steady-state mixing ratios of HO2 radicals ([HO2]ss) reach up to molec cm−3 when 16.2 ppm H2O2 is continuously injected into the chamber. If 110 ppb H2O2 is used instead, the resulting [HO2]ss levels fall close to the ambient range (∼108 molec cm−3).
Simulated steady-state NO mixing ratios in the chamber range from ∼2 ppt to ∼0.9 ppb from combinations of 0.1–20 ppb NO and 0.11–16.22 ppm H2O2 in the injection flow. The ratio of inflow NO concentration to the steady-state NO concentration in the chamber ranges from 5 to 93, demonstrating the importance of chemical removal in controlling the overall steady-state NO levels. O3 accumulation is an inevitable consequence under photolytic conditions and, for example, the presence of 10 ppb O3 leads to the chemical removal term () in Eq. (1) that reduces the steady-state NO concentration by a factor of 60. It is worth noting that under all simulated conditions in the continuous flow mode, O3 (∼1–126 ppb) coexists with NO (∼0.002–0.9 ppb). This particular chemical scenario, which is impossible to achieve in batch-mode reactors due to prompt conversion of NO to NO2, could then be used to mimic ambient ozonolysis chemistry, for example, in forest regions that frequently encounter polluted air masses from nearby urban areas. The steady-state mixing ratios of NO2 ([NO2]ss) exhibit a strong linear correlation with NO influxes. The use of less than 20 ppb NO in the injection flow results in a few to tens of ppb [NO2]ss, which is higher than the range typically found in the ambient. The potential “quenching” effect of NO2 on RO2 radicals through reversible termolecular reactions is discussed shortly.
In the so-called “high-NOx” chamber experiments, the NO3 radical is an unavoidable side-product when black lights are used as a representative of the solar radiation in mimicking the daytime photochemistry in the troposphere. The photolysis of NO3, although its primary sink in the atmosphere, proceeds rather slowly () under the present chamber photolytic conditions, thereby leading to a significant accumulation of NO3 radicals (7.9×104–2.8×108 molec cm−3) at steady state. The simulated NO3 ∕ OH ratio dictates the extent to which the NO3 (nighttime) chemistry competes with the OH-initiated (daytime) photochemistry. For compounds that are highly reactive towards NO3 such as isoprene, NO3-initiated oxidation accounts for up to ∼60 % of the overall isoprene degradation pathways at the highest NO3 ∕ OH ratio (∼255) simulated. Low concentrations of NO (<20 ppb) and H2O2 (<2 ppm) in the injection flow are therefore necessary to limit the interferences of NO3-initiated chemistry. Again, taking isoprene as an example, the NO3 oxidation pathway contributes less than 0.1 % of the overall isoprene degradation kinetics at the lowest NO3 ∕ OH ratio (∼0.13) simulated here.
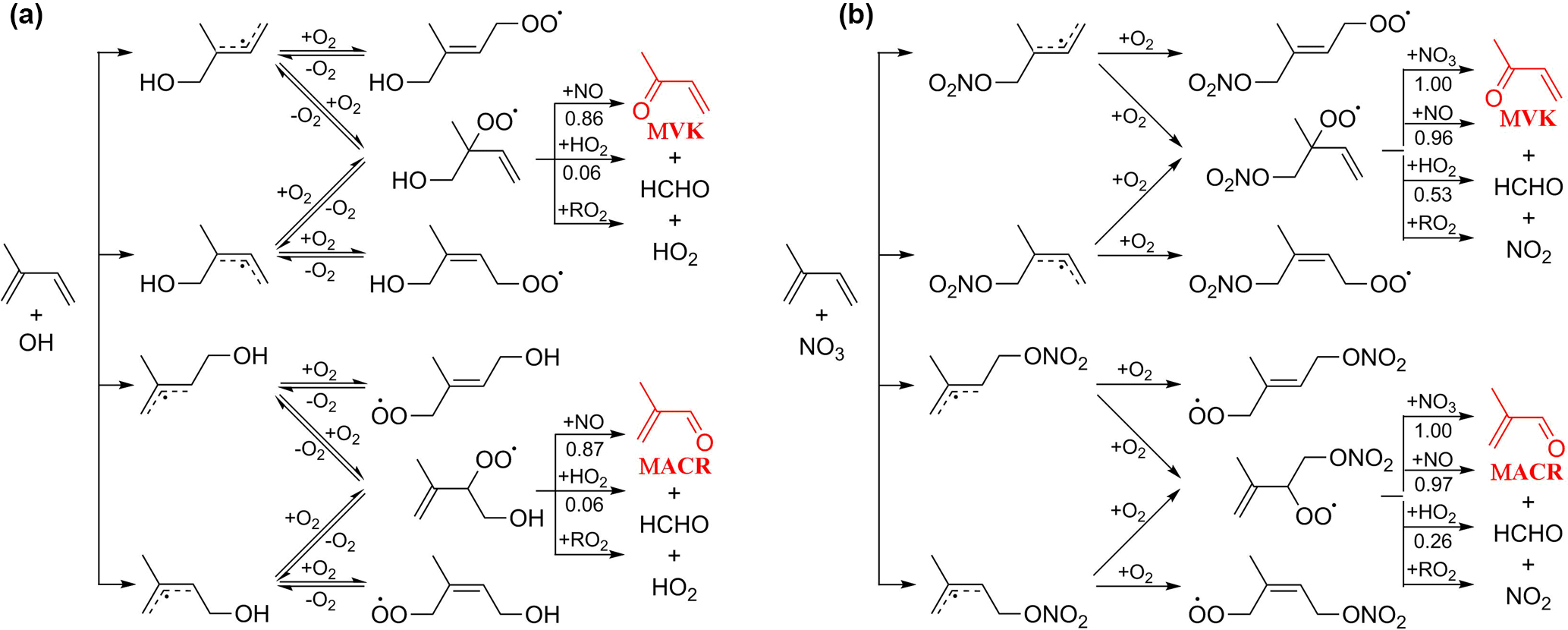
Figure 2Representative mechanism for (a) OH- and (b) NO3-initiated oxidation of isoprene that leads to the formation of MACR and MVK.
Also given in Fig. 1 is the calculated lifetime () of an RO2 radical with respect to reactions with NO and HO2 at 306 K. In most batch-mode chamber experiments, of only several seconds or less can be achieved due to the presence of tens to hundreds of ppb levels of NO and HO2. Here could extend to 60 s or even longer with the continuous input of low mixing ratios of H2O2 (≤110 ppb) and NO (≤0.2 ppb). Note that the presence of tens of ppb NO2 in the chamber might impose a “quenching” effect on the steady-state RO2 level through rapid reversible reactions (). We evaluate this potential “quenching effect” using ethyl peroxy radical (C2H5O2) generated from OH oxidation of ethane as an example. Simulations shown in Fig. S3 in the Supplement reveal that incorporation of the reaction into the mechanism in the presence of ∼1–80 ppb NO2 does not notably change the behavior of C2H5O2 radical. One exception is the peroxyacyl radical, which combines with NO2 yielding peroxyacyl nitrate. For example, under 0.1–16 ppb [NO2]ss as displayed in Fig. 1, we calculate that the time needed for peroxyacetyl radical (CH3C(O)O2) to reach equilibrium with peroxyacetyl nitrate () ranges from ∼ 1 to ∼ 10 s, suggesting that the lifetime of peroxyacyl radicals is ultimately controlled by NO2 instead of NO ∕ HO2 in the reaction system and, consequently, peroxyacyl radicals are not expected to be long-lived in the current chamber configuration.
We further compare the photochemical oxidation environment created here with the “intermediate-NO” conditions achieved by other chambers that employed the experimental approaches introduced earlier. In terms of the oxidizing power, all approaches are capable of maintaining an atmospheric relevant OH level (∼106 molec cm−3) except the “slow chemistry” scenario that limits the photolysis rate of the OH precursor and results in an average OH mixing ratio of ∼105 molec cm−3 (Crounse et al., 2012; Teng et al., 2017). At comparable OH levels, the overall atmospheric OH exposure achieved in the flow tube reactor is rather limited due to the short residence time (e.g., ∼80 s in the PAM reactor). In terms of the NOx level, precisely controlled steady-state NO concentration can be achieved for an indefinite time period by operating chambers in the continuously mixed flow mode. However, NO2 accumulates during the continuous oxidation process and the resulting NO2 ∕ NO ratio can be as much as an order of magnitude higher than that achieved in the static outdoor chambers.
4.2 Application to OH-initiated oxidation of isoprene
MACR and MVK are major first-generation products from the OH-initiated oxidation of isoprene in the presence of NO (Wennberg et al., 2018). They are produced from the decomposition of β-ISOPO alkoxy radicals that are primarily formed from the reaction of β-ISOPOO peroxy radicals (β-1-OH-2-OO and β-4-OH-3-OO) with NO; see mechanisms displayed in Fig. 2a. Reactions of β-ISOPOO peroxy radicals with HO2 and RO2 also partially yield β-ISOPO alkoxy radicals that ultimately lead to MACR and MVK, although these pathways are considered to be minor in the presence of hundreds to thousands of ppt NO in the atmosphere. The molar yields determined from previous studies range from 30 to 35 % for MVK and from 20 to 25 % for MACR under high-NO conditions (NO >60 ppb) (Tuazon and Atkinson, 1990; Paulson and Seinfeld, 1992; Miyoshi et al., 1994; Ruppert and Becker, 2000; Sprengnether et al., 2002; Galloway et al., 2011; Liu et al., 2013). It has been recently shown that the six hydroxyl peroxy radicals (ISOPOO) produced from the initial OH addition to the double bonds of isoprene undergo rapid interconversion by removal or addition of O2 that competes with bimolecular reactions under atmospherically relevant NO levels (Peeters et al., 2014; Teng et al., 2017). As a result, the distribution of ISOPOO radical isomers and their subsequent reaction products varies with their lifetimes with respect to bimolecular reactions. In the presence of hundreds of ppb NO as done by most previous experimental studies, the reaction of ISOPOO radicals with NO dominates over their interconversion, and the production of β-ISOPOO peroxy radical is less favored, leading the experiments to underestimate the MACR and MVK yields typically obtained in ambient conditions. Measurements by Karl et al. (2006) and Liu et al. (2013) conducted at NO concentrations comparable to the moderately polluted urban environment (∼0.2 ppb in Karl et al. and ∼1 ppb in Liu et al.) found higher MACR (∼27 % in Karl et al. and ∼31.8 % in Liu et al.) and MVK (∼41 % in Karl et al. and ∼44.5 % in Liu et al.) yields than other studies.
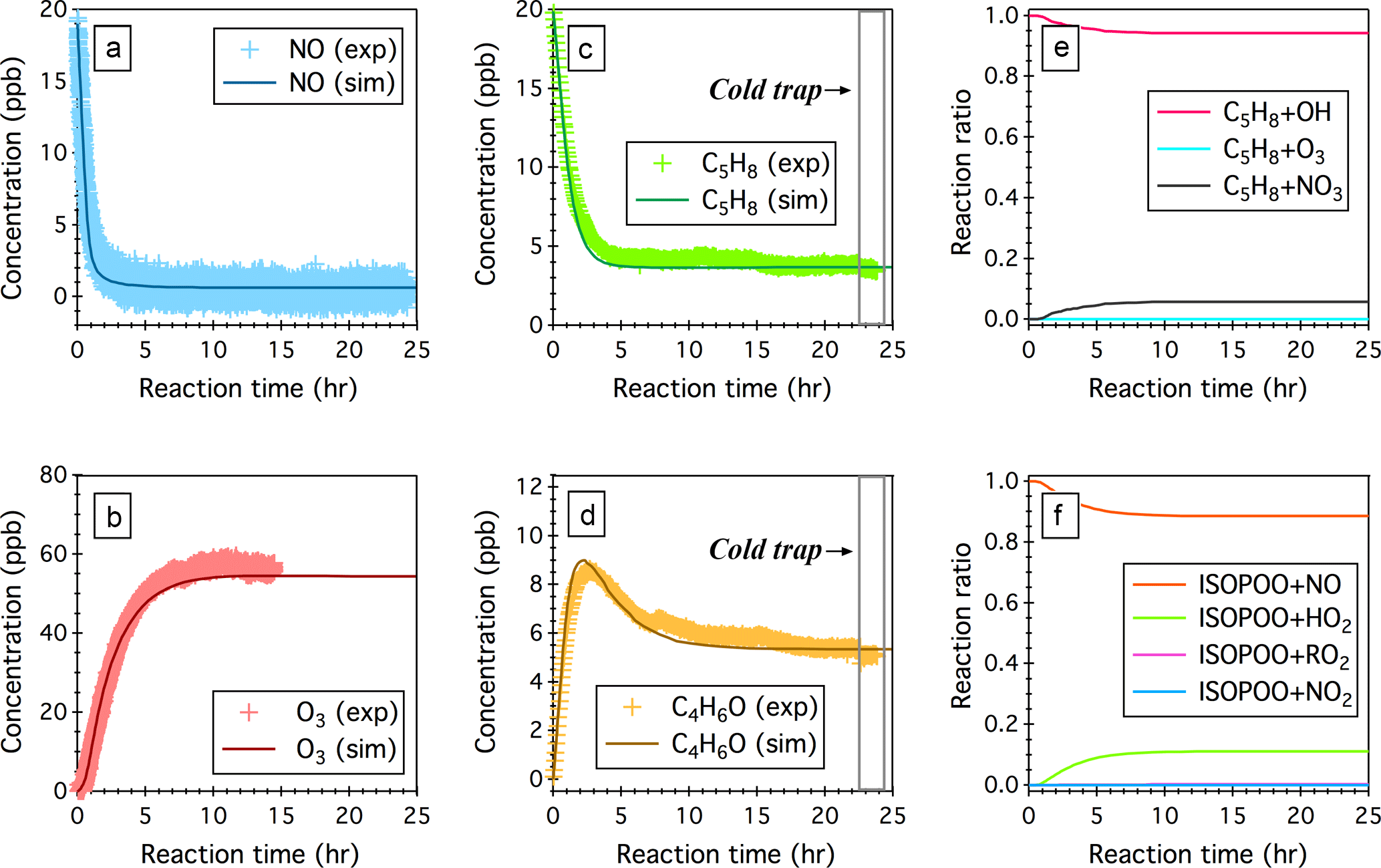
Figure 3Simulated (sim.) and observed (exp.) temporal profiles of (a) NO, (b) O3, (c) isoprene (C5H8), and (d) the sum of MACR and MVK (C4H6O) over 24 h OH-initiated oxidation of isoprene in the continuous flow mode chamber operation. Also displayed here include (e) simulated fractions of OH oxidation, ozonolysis, and NO3 oxidation as the removal pathways of isoprene and (f) simulated fractions of ISOPOO radicals that react with NO, HO2, RO2, and NO3. Time 0 is the point at which the chamber lights are turned on. Initial experimental conditions are 19 ppb NO, 0 ppb NO2, 0 ppb O3, 600 ppb H2O2, and 19.9 ppb C5H8, with continuous input of 600 ppb H2O2, 19 ppb NO, and 19.9 ppb C5H8 over the course of 24 h photochemical reactions.
Here we perform a steady-state continuous-mode experiment to measure the production of MACR and MVK from the OH-initiated oxidation of isoprene in the presence of ∼0.45 ppb NO. Figure 3 shows the observed and simulated temporal profiles of NOx, O3, C5H8, and C4H6O over 24 h photooxidation of isoprene. In this experiment, C5H8, H2O2, and NO were continuously fed into the chamber, with constant inflow concentrations of 19.9, 600, and 19 ppb, respectively. An outgoing flow at 40 L min−1 continuously withdrew air from the chamber to balance the pressure. After approaching steady state, the sampling tube was submerged into a low-temperature ethanol bath (−40 ± 2 ∘C) to trap oxidized products that would otherwise undergo thermal decomposition introducing interferences in the C4H7O+ (m∕z 71) signal. The measured concentrations of C5H8 and C4H6O upon cold trapping agree within 2.8 and 4.6 % uncertainties with the model simulations (see Fig. 3c and d).
To calculate the total molar yield () of MACR and MVK, two reactions are considered:
where k1 is the rate constant for OH reaction with isoprene, and k2 is taken as the average of rate constants for OH reactions with MACR and MVK. Uncertainties associated with the simplification of k2 in calculating the MACR and MVK yields will be discussed shortly. Note that the ozonolysis and NO3-initiated oxidation in total account for less than 6 % of isoprene degradation pathway under current experimental conditions and are neglected in the calculation. The ozonolysis and photolysis in total account for ∼6 % of the C4H6O degradation pathway and are neglected here as well.
In the continuous-mode operation, two mass conservation equations are satisfied at steady state:
where [C5H8]ss and [C4H6O]ss are the PTR-Q-MS measured steady-state concentrations of isoprene and the sum of MACR and MVK when using the cold trap, respectively, [C5H8]0 is the initial concentration of isoprene, and τ is the chamber mean residence time and can be calculated as the total chamber volume divided by the incoming and outgoing flow rate. The steady-state OH radical concentration ([OH]ss) can be derived by solving Eq. (2). The calculated [OH]ss (3.13×106 molec cm−3) is 12 % higher than the model prediction (2.74×106 molec cm−3). The molar yield of the sum of MACR and MVK from isoprene OH oxidation pathway in the presence of ∼0.45 ppb NO is thus given by Eq. (4) and calculated as 76.7 ± 5.8 %:
Here a 5.8 % uncertainty originates from the assumption that MACR + OH and MVK + OH proceed with equal reaction rate, although the rate constant for MVK + OH is ∼31 % lower than that of MACR + OH. Another potential uncertainty relates to the accuracy of the simulated steady-state HO2 and RO2 concentrations and the contribution of β-ISOPOO + HO2 and β-ISOPOO + RO2 reaction pathways to the overall β-ISOPOO fate. The fraction of β-ISOPOO radicals that reacts with NO (fβ-ISOPOO+NO) was predicted as 0.88, which was used here to scale the final MACR + MVK yield, see Eq. (4).
In summary, the measured yield of the sum of MACR and MVK in this study is close to that reported by Karl et al. (2006) and Liu et al. (2013), but ∼27–52 % higher than the majority of previous measurements performed under high-NO conditions (NO >60 ppb). This is consistent with the dynamic nature of the six ISOPOO radical isomers that undergo rapid interconversion by addition or removal of O2. In the presence of ∼0.45 ppb NO as performed in this study, lifetimes of the β-1-OH-2-OO peroxy radical with respect to reaction with NO and loss of O2 are estimated as 9.4 and 0.2 s, respectively, implying that the rapid interconversion between β-ISOPOO and δ-ISOPOO radicals essentially governs their distribution; under such conditions, the production of thermodynamically more stable β-ISOPOO isomers is favored, leading to higher yields of MACR and MVK. Here the reported MACR and MVK yield from isoprene OH oxidation in the presence of ∼0.45 ppb NO represents an illustration of chamber operation at steady-state continuous flow mode for the establishment of certain experimental conditions that are not easily accessible from traditional batch-mode chamber experiments. A complete measurement of first-generation oxidation products from isoprene OH reaction under a wide range of NO levels (ISOPOO bimolecular lifetimes) will be forthcoming in a future publication.
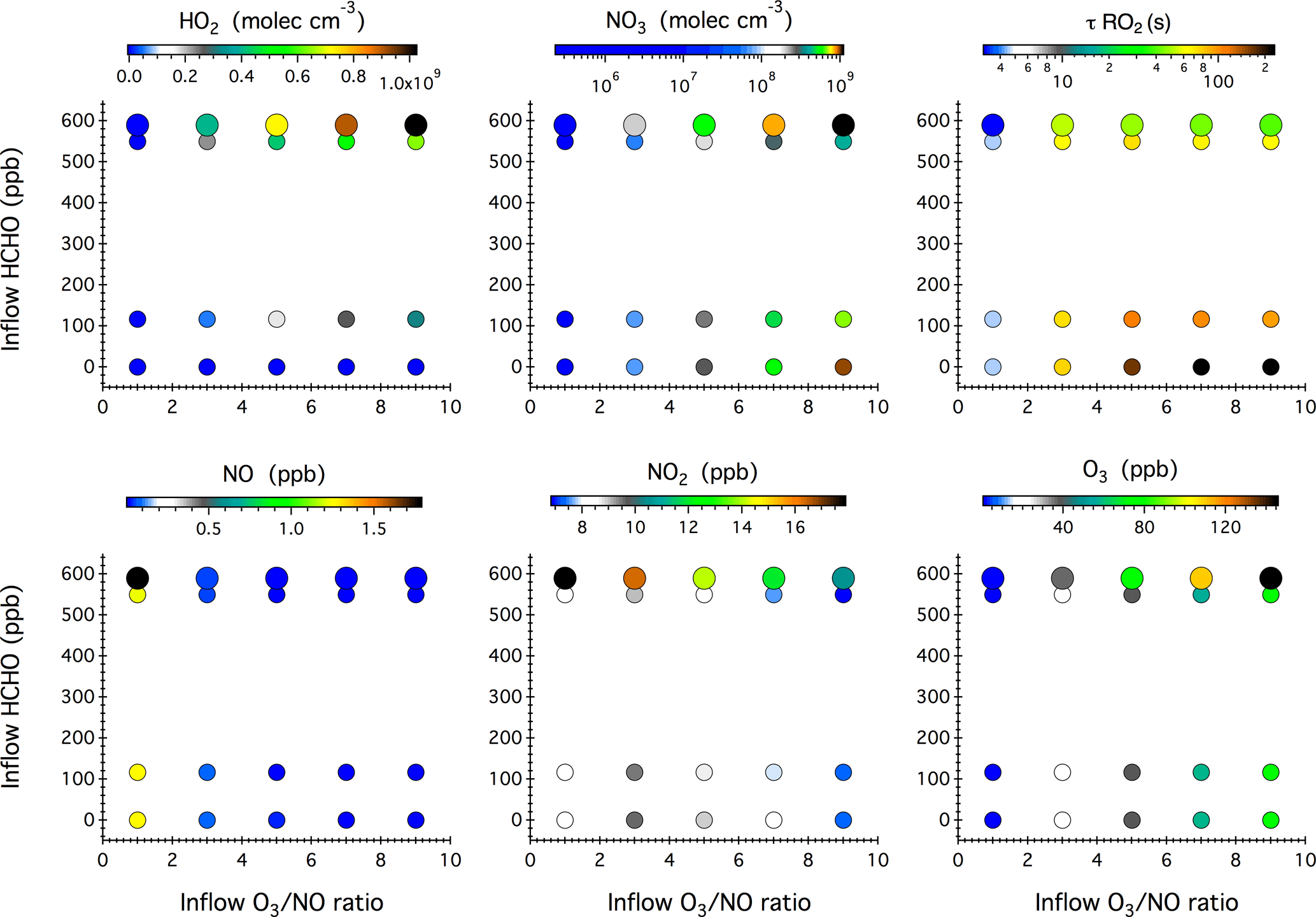
Figure 4Simulated steady-state mixing ratios of HO2, NO3, NO, NO2, and O3 after 16 h of dark reactions in the chamber as a function of the concentrations of HCHO, NO, and O3 in the continuous injection flow. The symbol size denotes different inflow NO concentrations, i.e., 10 and 20 ppb. Also given here is the calculated lifetime of an RO2 radical () with respect to reactions with NO, NO3, and HO2.
4.3 Optimal operating conditions for nighttime chemistry
Figure 4 shows the model simulated steady-state mixing ratios of HO2, NO3, NO, NO2, and O3 after 16 h of dark reactions in the chamber as a function of the HCHO concentration and O3 ∕ NO ratio in the continuous chamber inflow. Blank experiments were compared with simulations in five cases (see Table S1 in the Supplement). The model captures the evolution patterns of NOx and O3 well. The observed mixing ratios of NO2 and O3 agree with the simulations to within 11 and 6 %, respectively (Fig. S4 in the Supplement).
Compared with the photochemical reaction schemes discussed earlier, the nocturnal chemistry is rather straightforward; that is, the inflow O3 ∕ NO ratio governs the steady-state concentrations of NO3, NOx, and O3, while the inflow HCHO concentration ultimately controls the steady-state HO2 level. Increasing the O3 ∕ NO ratio from 1 to 9 in the continuous inflow leads to increased NO3 from 2.4×105 to 1.1×109 molec cm−3, but decreased NO from 1.8 ppb to 20 ppt and decreased NO2 from 18 to 7 ppb. At a fixed inflow O3 ∕ NO ratio, doubling the NO and O3 concentrations leads to elevated NO3, NOx, and O3 by a factor of 2.0–3.2, 1.5–2.0, and 1.4–2.0, respectively. The use of HCHO as an effective dark HO2 source does not significantly impact the steady-state mixing ratios of NOx and O3, but slightly weakens the NO3 production.
The calculated RO2 lifetime () with respect to reactions with NO, NO3, and HO2 at 295 K ranges from 3 to 225 s. The highest was achieved in the absence of any HCHO source and corresponds to a chemical regime that can be employed to study the intramolecular isomerization (autoxidation) pathway of RO2 radicals, if any. Adding a continuous flow of HCHO to the system leads to the production of 107–109 molec cm−3 HO2 radicals that then constitute a significant sink of RO2 radicals and represents prevailing forest environments during nighttime.
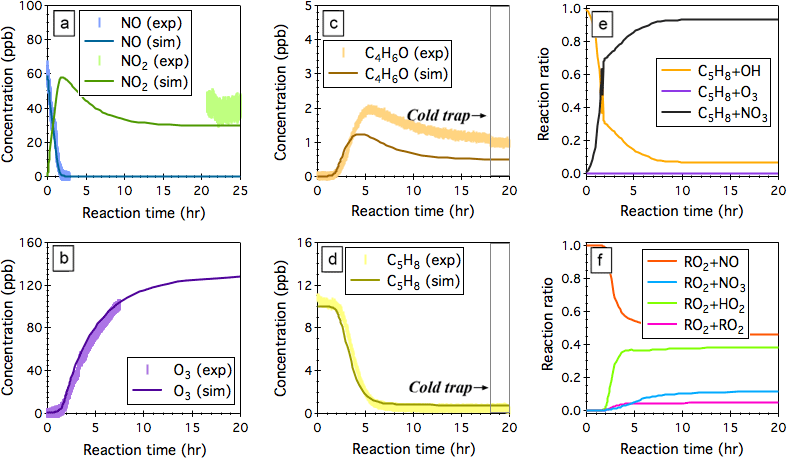
Figure 5Simulated (sim.) and observed (exp.) evolution patterns of (a) NOx, (b) O3, (c) the sum of MACR and MVK (C4H6O), and (d) isoprene (C5H8) over 20 h NO3-initiated oxidation of isoprene under continuous flow mode chamber operation. The fractions of isoprene that reacts with OH, O3, and NO3 are given in panel (e), and the fractions of INOO radical that undergoes bimolecular reactions with NO, NO3, HO2, and RO2 are given in panel (f). Initial experimental conditions are 0 ppb O3, 59 ppb NOx, and 10.2 ppb C5H8, with continuous input of 205 ppb O3, 59 ppb NO, and 10.2 ppb C5H8 over the course of 20 h dark reactions.
4.4 Application to NO3-initiated oxidation of isoprene
NO3-initiated oxidation of isoprene proceeds by the NO3 addition to the carbon double bonds followed by O2 addition, yielding six distinct nitrooxy peroxy radicals (INOO), including two isomers (β-INOO) with O2 added on the β-carbon to the nitrate group (see Fig. 2b for schematic illustration). The β-INOO radicals react further with NO3, HO2, NO, and RO2, producing nitrooxy alkoxy radicals (β-INO) with molar yields of 1.00, 0.53, 0.97, and 0.40, respectively (Wennberg et al., 2018). The further decomposition of β-INO radicals produces MACR and MVK, together with HCHO and NO2. Depending on the actual fate of β-INOO radicals, the yields of β-INO radicals can then vary from 0.4 to 1.0, resulting in a distinct distribution of final oxidation products. It is thus important to specify the ultimate fate of INOO radicals during quantification of oxidation products from isoprene reaction with NO3. As an illustration, we performed one continuous-mode experiment that targets on controlling the steady-state fate of INOO radicals to be their reaction with NO and HO2 (46 and 38 % INOO radicals are predicted to react with NO and HO2, respectively, as shown in Fig. 5). Note that by adjusting the concentrations and fractions of inflow reactants (O3, NO, HCHO, and C5H8), different chemical fates and lifetimes of INOO radicals can be achieved.
Figure 5 shows the observed and predicted temporal profiles of NOx, O3, C5H8, and C4H6O over 25 h of isoprene oxidation by NO3, with continuous input of 10.2 ppb C5H8, 205 ppb O3, and 59 ppb NO into the chamber and a balancing outgoing flow at 40 L min−1 carrying well-mixed reactants and products. It took >16 h to reach steady state for all the species displayed. In general, the model captures the trends of O3 and NO well, while underpredicting the steady-state NO2 by ∼26 %. After ∼18 h of dark reaction, the PTR-Q-MS sampling tubing was submerged into a cold ethanol bath (−40 ± 2 ∘C) to trap artifacts in the PTR-Q-MS measured C4H7O+ (m∕z 71) signal. The simulated steady-state concentration of isoprene agrees within 9 % with the measurements. The derived concentration of the sum of MACR and MVK from the measured C4H7O+ ion intensity upon cold trapping is ∼1.1 ppb, which is ∼129 % higher than the model predictions (∼0.48 ppb). This disagreement can be attributed, to a large extent, to the oversimplified representation of the six different INOO radicals as one δ-INOO isomer in the MCMv3.3.1 mechanism. As a result, the production of β-INOO radical, the important precursor of MACR and MVK, from NO3-initiated oxidation of isoprene is suppressed in the simulations. The measured molar yield of the sum of MACR and MVK is 36.3 ± 12.1 %, with uncertainties arising from the fact that 10.5 % isoprene is predicted to react with OH as an additional source of MACR and MVK. Using this value, the fraction of β-INOO over the sum of nitrooxy peroxy radicals is estimated as 48.6 ± 16.2 %, which is close to that (∼46.3 %) reported by Schwantes et al. (2015), although the estimated bimolecular lifetime of INOO radical in that study (∼30 s) is lower than that predicted in the present work (∼50 s). As discussed above, the hydroxyl peroxy radicals produced from OH oxidation of isoprene could undergo rapid interconversion through addition or removal of O2 at atmospherically relevant lifetimes. This interconversion significantly impacts the subsequent chemistry of individual ISOPOO radical isomers in terms of reaction rates and product distributions. It is likely that the INOO radicals follow similar interconversion due to the small R-OO bond dissociation energy, although no experimental evidence exists. A full examination of the INOO chemistry, i.e., their kinetic and thermodynamic properties as well as their chemical fate at different lifetimes, will be the focus of future studies using this continuous flow chamber operation method.
We report here the development and characterization of the NCAR Atmospheric Simulation Chamber operated at steady-state continuous flow mode for simulating daytime and nocturnal chemistry under atmospherically relevant NO levels. The chamber is designed to achieve a well-controlled steady-state environment by continuous inflow of reactants and continuous withdrawal of reactor contents. We use a combination of kinetic modeling and chamber experiments to characterize the “intermediate-NO” chemical regime (tens of ppt to a few ppb) that can be achieved by precisely controlling the inlet reactant concentrations and the mixing/residence timescales of the chamber.
To mimic daytime photochemistry, continuous input of H2O2 and NO gases is required, resulting in steady-state OH mixing ratios of 105–106 molec cm−3 under irradiation. Under such conditions, the lifetime of a peroxy radical with respect to reaction with NO and HO2 can be extended to 60 s or even longer, thus providing a unique environment to study all reaction possibilities of RO2 radicals including the intramolecular isomerization (autoxidation) pathway. When studying OH-initiated chemistry, care needs to be taken to avoid a range of experimental conditions (e.g., inflow H2O2 >2 ppm and NO>20 ppb) where NO3 oxidation might account for a large fraction of the overall degradation pathway of certain parent hydrocarbons such as alkenes.
To mimic nighttime chemistry, continuous input of NO (or NO2) and O3 is needed to produce steady-state NO3 radicals in the range of 106–109 molec cm−3 in the dark. Under such conditions, an RO2 radical can live up to 4 min prior to finding a bimolecular reaction partner (e.g., NO, NO3, and HO2), which were the dominant fates of RO2 radicals in most batch-mode chamber experiments. Again, the long lifetime of RO2 radicals achieved by the steady-state continuous-mode operation opens an avenue for close examination of RO2 unimolecular (isomerization) pathways in nocturnal environments.
In simulating both daytime and nighttime chemistry with continuous flow operation method, O3 accumulation is unavoidable. The extent to which ozonolysis interferes with OH- or NO3-initiated oxidation chemistry depends on the steady-state O3 concentration achieved in the chamber and its reactivity towards various parent VOCs. Taking isoprene as an example, ozonolysis accounts for <1 and <0.1 % of the overall isoprene degradation kinetics, respectively, under established steady-state photolytic and dark conditions described above.
In atmospheric chemistry, the terms “zero-NO” vs. “high-NO” have been widely used to classify photooxidation conditions and delineate the gas-phase fate of the peroxy radicals (RO2) generated from VOCs oxidation (Zhang et al., 2010, 2014b, 2015a; He et al., 2011; Cappa et al., 2013; Zhang and Seinfeld, 2013; Loza et al., 2014; Nguyen et al., 2014a; Schilling Fahnestock et al., 2014; Krechmer et al., 2015; Gordon et al., 2016; Riva et al., 2016; Thomas et al., 2016; Schwantes et al., 2017a, b). In the so-called “high-NO” regime, reaction with NO dominates the fate of RO2 radicals, whereas in the “zero-NO” regime, the RO2 radicals primarily undergo reaction with HO2 and, perhaps to a much lesser degree, self- or cross-combination. The importance of the “intermediate-NO” regime lies in the fact that at sub-ppb levels of NO, the RO2+NO and RO2+HO2 reactions are expected to co-exist and the RO2 radical could survive up to several minutes before encountering a partner (NO ∕ HO2) for bimolecular reactions. Under such conditions, the RO2 radical isomers may undergo interconversion by addition or removal of O2 and intramolecular isomerization (autoxidation) through H-shift. Here we use isoprene as an illustrative VOC to explore the fate of RO2 radicals under sub-ppb NO. Future work will focus on detailed characterization of oxidation products from isoprene day- and nighttime chemistry with particular attention given to the controlled RO2 fates and lifetimes.
Data presented in this paper are available upon request to the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/amt-11-2537-2018-supplement.
The authors declare that they have no conflict of interest.
The National Center for Atmospheric Research is operated by the University
Corporation for Atmospheric Research, under the sponsorship of the National
Science Foundation.
Edited by: Andreas Hofzumahaus
Reviewed by: Paul Seakins and one anonymous referee
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006.
Bloss, C., Wagner, V., Bonzanini, A., Jenkin, M. E., Wirtz, K., Martin-Reviejo, M., and Pilling, M. J.: Evaluation of detailed aromatic mechanisms (MCMv3 and MCMv3.1) against environmental chamber data, Atmos. Chem. Phys., 5, 623–639, https://doi.org/10.5194/acp-5-623-2005, 2005.
Cappa, C. D., Zhang, X., Loza, C. L., Craven, J. S., Yee, L. D., and Seinfeld, J. H.: Application of the Statistical Oxidation Model (SOM) to Secondary Organic Aerosol formation from photooxidation of C12 alkanes, Atmos. Chem. Phys., 13, 1591–1606, https://doi.org/10.5194/acp-13-1591-2013, 2013.
Chameides, W. L., Lindsay, R. W., Richardson, J., and Kiang, C. S.: The role of biogenic hydrocarbons in urban photochemical smog: Atlanta as a case study, Science, 241, 1473–1476, 1988.
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, 2011.
Crounse, J. D., Knap, H. C., Ørnsø, K. B., Jørgensen, S., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric fate of methacrolein. 1. Peroxy radical isomerization following addition of OH and O2, J. Phys. Chem. A, 116, 5756–5762, 2012.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, 2013.
De Gouw, J. A., Middlebrook, A. M., Warneke, C., Goldan, P. D., Kuster, W. C., Roberts, J. M., Fehsenfeld, F. C., Worsnop, D. R., Canagaratna, M. R., and Pszenny, A. A. P.: Budget of organic carbon in a polluted atmosphere: Results from the New England Air Quality Study in 2002, J. Geophys. Res.-Atmos., 110, D16305, https://doi.org/10.1029/2004JD005623, 2005.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476, https://doi.org/10.1038/nature13032, 2014.
EPA: Data from the 2011 National Emissions Inventory, Version 1, available at: http://www.epa.gov/air-emissions-inventories/2011-national-emissions-inventory-nei-data, last access: 2014.
Galloway, M. M., Huisman, A. J., Yee, L. D., Chan, A. W. H., Loza, C. L., Seinfeld, J. H., and Keutsch, F. N.: Yields of oxidized volatile organic compounds during the OH radical initiated oxidation of isoprene, methyl vinyl ketone, and methacrolein under high-NOx conditions, Atmos. Chem. Phys., 11, 10779–10790, https://doi.org/10.5194/acp-11-10779-2011, 2011.
Goldstein, A. H., Koven, C. D., Heald, C. L., and Fung, I. Y.: Biogenic carbon and anthropogenic pollutants combine to form a cooling haze over the southeastern United States, P. Natl. Acad. Sci. USA, 106, 8835–8840, 2009.
Gordon, H., Sengupta, K., Rap, A., Duplissy, J., Frege, C., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Dunne, E. M., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J. S., Dias, A., Ehrhart, S., Fischer, L., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Kirkby, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Monks, S. A., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K. J., Richards, N. A. D., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sharma, S., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Curtius, J., and Carslaw, K. S.: Reduced anthropogenic aerosol radiative forcing caused by biogenic new particle formation, P. Natl. Acad. Sci. USA, 113, 12053–12058, 2016.
He, S., Chen, Z., and Zhang, X.: Photochemical reactions of methyl and ethyl nitrate: a dual role for alkyl nitrates in the nitrogen cycle, Environ. Chem., 8, 529–542, 2011.
Huang, D. D., Zhang, X., Dalleska, N. F., Lignell, H., Coggon, M. M., Chan, C. M., Flagan, R. C., Seinfeld, J. H., and Chan, C. K.: A note on the effects of inorganic seed aerosol on the oxidation state of secondary organic aerosol – α-pinene ozonolysis, J. Geophys. Res.-Atmos., 121, 12476–12483, 2016.
Huang, Y., Zhao, R., Charan, S. M., Kenseth, C. M., Zhang, X., and Seinfeld, J. H.: Unified theory of vapor-wall mass transport in Teflon-walled environmental chambers, Environ. Sci. Technol., 52, 2134–2142, 2018.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., and Worsnop, D. R.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, 2015.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Karl, M., Dorn, H. P., Holland, F., Koppmann, R., Poppe, D., Rupp, L., Schaub, A., and Wahner, A.: Product study of the reaction of OH radicals with isoprene in the atmosphere simulation chamber SAPHIR, J. Atmos. Chem., 55, 167–187, 2006.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., João, A., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, 2016.
Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., III, R. L. M., Stark, H., Jayne, J. T., Sipila, M., Junninen, H., Clair, J. M. S., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R.: Formation of low volatility organic compounds and secondary organic aerosol from isoprene hydroxyhydroperoxide low-NO oxidation, Environ. Sci. Technol., 49, 10330–10339, 2015.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of gas-wall partitioning in Teflon environmental chambers using rapid bursts of low-volatility oxidized species generated in situ, Environ. Sci. Technol., 50, 5757–5765, 2016.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, 2008.
Kurtén, T., Rissanen, M. P., Mackeprang, K., Thornton, J. A., Hyttinen, N., Jørgensen, S., Ehn, M., and Kjaergaard, H. G.: Computational study of hydrogen shifts and ring-opening mechanisms in α-pinene ozonolysis products, J. Phys. Chem. A, 119, 11366–11375, 2015.
Lambe, A., Massoli, P., Zhang, X., Canagaratna, M., Nowak, J., Daube, C., Yan, C., Nie, W., Onasch, T., Jayne, J., Kolb, C., Davidovits, P., Worsnop, D., and Brune, W.: Controlled nitric oxide production via O(1D) + N2O reactions for use in oxidation flow reactor studies, Atmos. Meas. Tech., 10, 2283–2298, https://doi.org/10.5194/amt-10-2283-2017, 2017.
Liu, Y. J., Herdlinger-Blatt, I., McKinney, K. A., and Martin, S. T.: Production of methyl vinyl ketone and methacrolein via the hydroperoxyl pathway of isoprene oxidation, Atmos. Chem. Phys., 13, 5715–5730, https://doi.org/10.5194/acp-13-5715-2013, 2013.
Loza, C. L., Craven, J. S., Yee, L. D., Coggon, M. M., Schwantes, R. H., Shiraiwa, M., Zhang, X., Schilling, K. A., Ng, N. L., Canagaratna, M. R., Ziemann, P. J., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol yields of 12-carbon alkanes, Atmos. Chem. Phys., 14, 1423–1439, https://doi.org/10.5194/acp-14-1423-2014, 2014.
McVay, R. C., Zhang, X., Aumont, B., Valorso, R., Camredon, M., La, Y. S., Wennberg, P. O., and Seinfeld, J. H.: SOA formation from the photooxidation of α-pinene: systematic exploration of the simulation of chamber data, Atmos. Chem. Phys., 16, 2785–2802, https://doi.org/10.5194/acp-16-2785-2016, 2016.
Miyoshi, A., Hatakeyama, S., and Washida, N.: OH radical-initiated photooxidation of isoprene: An estimate of global CO production, J. Geophys. Res.-Atmos., 99, 18779–18787, 1994.
Nah, T., McVay, R. C., Zhang, X., Boyd, C. M., Seinfeld, J. H., and Ng, N. L.: Influence of seed aerosol surface area and oxidation rate on vapor wall deposition and SOA mass yields: a case study with α-pinene ozonolysis, Atmos. Chem. Phys., 16, 9361–9379, https://doi.org/10.5194/acp-16-9361-2016, 2016.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007.
Nguyen, T. B., Coggon, M. M., Bates, K. H., Zhang, X., Schwantes, R. H., Schilling, K. A., Loza, C. L., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 14, 3497–3510, https://doi.org/10.5194/acp-14-3497-2014, 2014a.
Nguyen, T. B., Crounse, J. D., Schwantes, R. H., Teng, A. P., Bates, K. H., Zhang, X., St. Clair, J. M., Brune, W. H., Tyndall, G. S., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): mechanistic chamber studies on the oxidation of biogenic compounds, Atmos. Chem. Phys., 14, 13531–13549, https://doi.org/10.5194/acp-14-13531-2014, 2014b.
Nguyen, T. B., Bates, K. H., Crounse, J. D., Schwantes, R. H., Zhang, X., Kjaergaard, H. G., Surratt, J. D., Lin, P., Laskin, A., and Seinfeld, J. H.: Mechanism of the hydroxyl radical oxidation of methacryloyl peroxynitrate (MPAN) and its pathway toward secondary organic aerosol formation in the atmosphere, Phys. Chem. Chem. Phys., 17, 17914–17926, 2015.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, 2012.
Paulson, S. E. and Seinfeld, J. H.: Development and evaluation of a photooxidation mechanism for isoprene, J. Geophys. Res.-Atmos., 97, 20703–20715, 1992.
Peeters, J., Müller, J.-F. o., Stavrakou, T., and Nguyen, V. S.: Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: The upgraded LIM1 mechanism, J. Phys. Chem. A, 118, 8625–8643, 2014.
Petropavlovskikh, I., Shetter, R., Hall, S., Ullmann, K., and Bhartia, P. K.: Algorithm for the charge-coupled-device scanning actinic flux spectroradiometer ozone retrieval in support of the Aura satellite validation, J. Appl. Remote Sens., 1, 013540, https://doi.org/10.1117/1.2802563, 2007.
Riva, M., Budisulistiorini, S. H., Chen, Y., Zhang, Z., D'Ambro, E. L., Zhang, X., Gold, A., Turpin, B. J., Thornton, J. A., and Canagaratna, M. R.: Chemical characterization of secondary organic aerosol from oxidation of isoprene hydroxyhydroperoxides, Environ. Sci. Technol., 50, 9889–9899, 2016.
Rivera-Rios, J. C., Nguyen, T. B., Crounse, J. D., Jud, W., St Clair, J. M., Mikoviny, T., Gilman, J. B., Lerner, B. M., Kaiser, J. B., and Gouw, J. d.: Conversion of hydroperoxides to carbonyls in field and laboratory instrumentation: Observational bias in diagnosing pristine versus anthropogenically controlled atmospheric chemistry, Geophys. Res. Lett., 41, 8645–8651, 2014.
Rollins, A. W., Browne, E. C., Min, K. E., Pusede, S. E., Wooldridge, P. J., Gentner, D. R., Goldstein, A. H., Liu, S., Day, D. A., and Russell, L. M.: Evidence for NOx control over nighttime SOA formation, Science, 337, 1210–1212, 2012.
Ruppert, L. and Becker, K. H.: A product study of the OH radical-initiated oxidation of isoprene: Formation of C 5-unsaturated diols, Atmos. Environ., 34, 1529–1542, 2000.
Schilling Fahnestock, K. A., Yee, L. D., Loza, C. L., Coggon, M. M., Schwantes, R., Zhang, X., Dalleska, N. F., and Seinfeld, J. H.: Secondary organic aerosol composition from C12 alkanes, J. Phys. Chem. A, 119, 4281–4297, 2014.
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St. Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 oxidation products from the RO2 + HO2 Pathway, J. Phys. Chem. A, 119, 10158–10171, 2015.
Schwantes, R. H., McVay, R. C., Zhang, X., Coggon, M. M., Lignell, H., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Science of the environmental chamber, Adv. Atmos. Chem., 1, 1–93, 2017a.
Schwantes, R. H., Schilling, K. A., McVay, R. C., Lignell, H., Coggon, M. M., Zhang, X., Wennberg, P. O., and Seinfeld, J. H.: Formation of highly oxygenated low-volatility products from cresol oxidation, Atmos. Chem. Phys., 17, 3453–3474, https://doi.org/10.5194/acp-17-3453-2017, 2017b.
Shilling, J. E., Chen, Q., King, S. M., Rosenoern, T., Kroll, J. H., Worsnop, D. R., McKinney, K. A., and Martin, S. T.: Particle mass yield in secondary organic aerosol formed by the dark ozonolysis of α-pinene, Atmos. Chem. Phys., 8, 2073–2088, https://doi.org/10.5194/acp-8-2073-2008, 2008.
Shilling, J. E., Zaveri, R. A., Fast, J. D., Kleinman, L., Alexander, M. L., Canagaratna, M. R., Fortner, E., Hubbe, J. M., Jayne, J. T., Sedlacek, A., Setyan, A., Springston, S., Worsnop, D. R., and Zhang, Q.: Enhanced SOA formation from mixed anthropogenic and biogenic emissions during the CARES campaign, Atmos. Chem. Phys., 13, 2091–2113, https://doi.org/10.5194/acp-13-2091-2013, 2013.
Singh, H. B. and Hanst, P. L.: Peroxyacetyl nitrate (PAN) in the unpolluted atmosphere: An important reservoir for nitrogen oxides, Geophys. Res. Lett., 8, 941–944, 1981.
Sprengnether, M., Demerjian, K. L., Donahue, N. M., and Anderson, J. G.: Product analysis of the OH oxidation of isoprene and 1, 3-butadiene in the presence of NO, J. Geophys. Res.-Atmos., 107, 4268, https://doi.org/10.1029/2001JD000716, 2002.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, 2010.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene peroxy radical dynamics, J. Am. Chem. Soc., 139, 5367–5377, 2017.
Thomas, D. A., Coggon, M. M., Lignell, H., Schilling, K. A., Zhang, X., Schwantes, R. H., Flagan, R. C., Seinfeld, J. H., and Beauchamp, J. L.: Real-time studies of iron oxalate-mediated oxidation of glycolaldehyde as a model for photochemical aging of aqueous tropospheric aerosols, Environ. Sci. Technol., 50, 12241–12249, 2016.
Tuazon, E. C. and Atkinson, R.: A product study of the gas-phase reaction of Isoprene with the OH radical in the presence of NOx, Int. J. Chem. Kinet., 22, 1221–1236, 1990.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-phase reactions of isoprene and its major oxidation products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., and Knote, C.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, 2015.
Zhang, X. and Seinfeld, J. H.: A functional group oxidation model (FGOM) for SOA formation and aging, Atmos. Chem. Phys., 13, 5907–5926, https://doi.org/10.5194/acp-13-5907-2013, 2013.
Zhang, X., Chen, Z. M., and Zhao, Y.: Laboratory simulation for the aqueous OH-oxidation of methyl vinyl ketone and methacrolein: significance to the in-cloud SOA production, Atmos. Chem. Phys., 10, 9551–9561, https://doi.org/10.5194/acp-10-9551-2010, 2010.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, 2014a.
Zhang, X., Schwantes, R. H., Coggon, M. M., Loza, C. L., Schilling, K. A., Flagan, R. C., and Seinfeld, J. H.: Role of ozone in SOA formation from alkane photooxidation, Atmos. Chem. Phys., 14, 1733–1753, https://doi.org/10.5194/acp-14-1733-2014, 2014b.
Zhang, X., McVay, R. C., Huang, D. D., Dalleska, N. F., Aumont, B., Flagan, R. C., and Seinfeld, J. H.: Formation and evolution of molecular products in α-pinene secondary organic aerosol, P. Natl. Acad. Sci. USA, 112, 14168–14173, 2015a.
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015b.
Zhang, X., Lambe, A. T., Upshur, M. A., Brooks, W. A., Gray Beì, A., Thomson, R. J., Geiger, F. M., Surratt, J. D., Zhang, Z., and Gold, A.: Highly oxygenated multifunctional compounds in α-pinene secondary organic aerosol, Environ. Sci. Technol., 51, 5932–5940, 2017.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, 2012.
intermediate NOregime characterized by a constant steady-state NO level ranging from tens of ppt to a few ppb in the chamber.