the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Feb 2019
| 22 Feb 2019
Large-volume air sample system for measuring 34S∕32S isotope ratio of carbonyl sulfide
Kazuki Kamezaki
Shohei Hattori
Enno Bahlmann
Naohiro Yoshida
Knowledge related to sulfur isotope ratios of carbonyl sulfide (OCS or COS), the most abundant atmospheric sulfur species, remains scarce. An earlier method developed for sulfur isotopic analysis for OCS using S+ fragmentation by an isotope ratio mass spectrometer is inapplicable for ambient air samples because of the large samples required (approx. 500 L of 500 pmol mol−1 OCS). To overcome this difficulty, herein we present a new sampling system for collecting approximately 10 nmol of OCS from ambient air coupled with a purification system. Salient system features are (i) accommodation of samples up to 500 L (approx. 10 nmol) of air at 5 L min−1; (ii) portability of adsorption tubes (1∕4 in. (0.64 cm) outer diameter, 17.5 cm length, approximately 1.4 cm3 volume) for preserving the OCS amount and δ34S(OCS) values at −80 ∘C for up to 90 days and 14 days; and (iii) purification OCS from other compounds such as CO2. We tested the OCS collection efficiency of the systems and sulfur isotopic fractionation during sampling. Results show precision (1σ) of δ34S(OCS) values as 0.4 ‰ for overall procedures during measurements for atmospheric samples. Additionally, this report presents diurnal variation of δ34S(OCS) values collected from ambient air at the Suzukakedai campus of the Tokyo Institute of Technology located in Yokohama, Japan. The observed OCS concentrations and δ34S(OCS) values were, respectively, 447–520 pmol mol−1 and from 10.4 ‰ to 10.7 ‰ with a lack of diurnal variation. The observed δ34S(OCS) values in ambient air differed greatly from previously reported values of δ34S(OCS) = (4.9±0.3) ‰ for compressed air collected at Kawasaki, Japan, presumably because of degradation of OCS in cylinders and collection processes for that sample. The difference of atmospheric δ34S(OCS) values between 10.5 ‰ in Japan (this study) and ∼13 ‰ recently reported in Israel or the Canary Islands indicates that spatial and temporal variation of δ34S(OCS) values is expected due to a link between anthropogenic activities and OCS cycles. The system presented herein is useful for application of δ34S(OCS) for investigation of OCS sources and sinks in the troposphere to elucidate its cycle.
- Article
(2288 KB) -
Supplement
(67 KB) - BibTeX
- EndNote





Carbonyl sulfide (OCS) is the most abundant sulfur-containing gas in ambient air with atmospheric concentrations of approximately 500 pmol mol−1 in the troposphere (Chin and Davis, 1995; Montzka et al., 2007). In fact, OCS can be transported to the stratosphere because the average residence time of OCS is longer than 2 years (Brühl et al., 2012). In the stratosphere, it is converted to stratospheric sulfate aerosols (SSAs) through atmospheric sink reactions (Crutzen, 1976). Therefore, OCS must be regarded as an important sulfur source for SSAs, playing an important role in the Earth's radiation budget and in ozone depletion. Moreover, because leaves consume OCS whenever assimilating CO2 but do not emit OCS to the atmosphere by respiration (Sandoval-Soto et al., 2005), OCS can be a tracer of gross primary production (GPP) on land (Campbell et al., 2008). For those reasons, elucidating the OCS dynamics in the atmosphere is important to elucidate the carbon cycle. Nevertheless, tropospheric OCS sources and sinks entail great uncertainty (Watts, 2000; Kremser et al., 2015) because of missing sources in the atmospheric budget of 230–800 Gg a−1 S equivalents as revealed by top-down modelling (Berry et al., 2013; Glatthor et al., 2015; Kuai et al., 2014).
Isotope analysis is a useful tool to trace sources and transformations of trace gases (Johnson et al., 2002; Brenninkmeijer, 2003). To quantify OCS sources and sinks in natural environments using isotope analysis, determination of isotopic fractionation for reactions and ambient measurements is required. To date, isotopic fractionations occurring in the reactions of OCS have been determined for almost all OCS sink reactions in the stratosphere: OCS photolysis (Hattori et al., 2011; Lin et al., 2011; Schmidt et al., 2013) as well as reactions with OH (Schmidt et al., 2012) and O(3P) (Hattori et al., 2012). Furthermore, the sulfur isotopic fractionation during soil bacterial degradation and enzymatic degradation was ascertained based on laboratory experiments (Kamezaki et al., 2016; Ogawa et al., 2017). Based on the analysis of commercially available compressed air, our group suggested a δ34S value of (4.9±0.3) ‰ for tropospheric OCS (Hattori et al., 2015). However, very recently, Angert et al. (2019) reported a markedly different δ34S value of (13.1±0.7) ‰ for tropospheric OCS using a gas chromatograph (GC) – multi-collector inductively coupled plasma mass spectrometer (MC–ICP-MS). For the measurement of sulfur isotope ratios (δ33S, δ34S, and Δ33S values) of OCS in our laboratory, an online method measuring on a GC–isotope ratio (IR)–MS using S+ fragmentation ions had been developed (Hattori et al., 2015). This method supports simple analysis of sulfur isotopic compositions of OCS over 8 nmol. However, application of this method for atmospheric samples has yet to be carried out using this GC–IR-MS method because of the large sample amounts that are necessary (i.e. 500 L of 500 pmol mol−1 OCS). Therefore, we aimed to develop a large-volume air sampling system to apply the S+ IR-MS method for atmospheric samples.
To date, several methods have been developed for concentration measurements using grab samples of air coupled with sampling–purification systems in the laboratory (e.g. Inomata et al., 1999; Xu et al., 2002; Montzka et al., 2004; Kato et al., 2012). Most systems collect 2–5 L of atmospheric samples for measuring OCS concentrations. The collected OCS is extracted in the laboratory with a combination of adsorbents at sub-ambient temperatures: Tenax TA with dry ice–methanol (Inomata et al., 1999) or dry ice–ethanol (Hattori et al., 2015), glass beads with liquid oxygen (Montzka et al., 2004) or liquid argon (Xu et al., 2002), or 2,3-tris (2-cyanoethoxy) propane with liquid oxygen (Kato et al., 2012). Grab sampling, however, is unrealistic when collecting 500 L of air. Therefore, we developed a large-volume air sampling system for measuring sulfur isotope ratios of OCS. We modified a large-volume air sampling system developed for carbon isotope measurement for halocarbons such as chloromethane and bromomethane, which have concentrations at pmol mol−1 levels in ambient air (Bahlmann et al., 2011). Subsequently, we combined this sampling system and newly developed an online OCS purification system for separation from impurities such as CO2, which is 106 times more abundant in air than OCS. For the current study, we describe the systems and its applications to atmospheric observation. We provide first results for diurnal variations of δ34S(OCS) in ambient air from samples collected at the Suzukakedai campus of the Tokyo Institute of Technology located in Yokohama, Japan.
2.1 Samples
An overview of the synthetic samples used for method evaluation in this study is given in Table 1. Commercial samples containing 10.5 % OCS balanced with high-purity He as sample A (99.99995 % purity; Japan Fine Products Co. Ltd., Kawasaki, Japan) and 5.9 µmol mol−1 OCS balanced with high-purity He as sample B (99.99995 % purity; Japan Fine Products Co. Ltd.) were used (Table 1). Furthermore, we synthesized OCS from three kinds of sulfur powders, designated as sample C produced from sulfur power (99.99 % purity; Fujifilm Wako Pure Chemical Corp., Japan), sample D produced from sulfur powder (99.98 % purity; Sigma-Aldrich Corp. LLC, Missouri, USA), and sample E (a mixture of sulfur powders used for samples C and D) with a reaction with CO (99.99 % purity; Japan Fine Products Co. Ltd., Kawasaki, Japan) in a manner similar to that described by Ferm (1957) and Hattori et al. (2015) (Table 1). The OCS concentrations for samples A and B were determined against the in-house synthesized OCS (i.e. 100 %) diluted to 10 % using high-purity He (99.99995 % purity; Japan Fine Products Co. Ltd.). It is noteworthy that the OCS concentration in sample B had showed no change at least 4 years after the publication of Hattori et al. (2015).
Table 1OCS samples balanced with He and synthesized OCS sample of averages and standard deviations (1σ) for sulfur isotope ratios for OCS measured for this study (sampling–purification system with GC–IR-MS) and with a conventional syringe injection system with GC–IR-MS as described by Hattori et al. (2015).
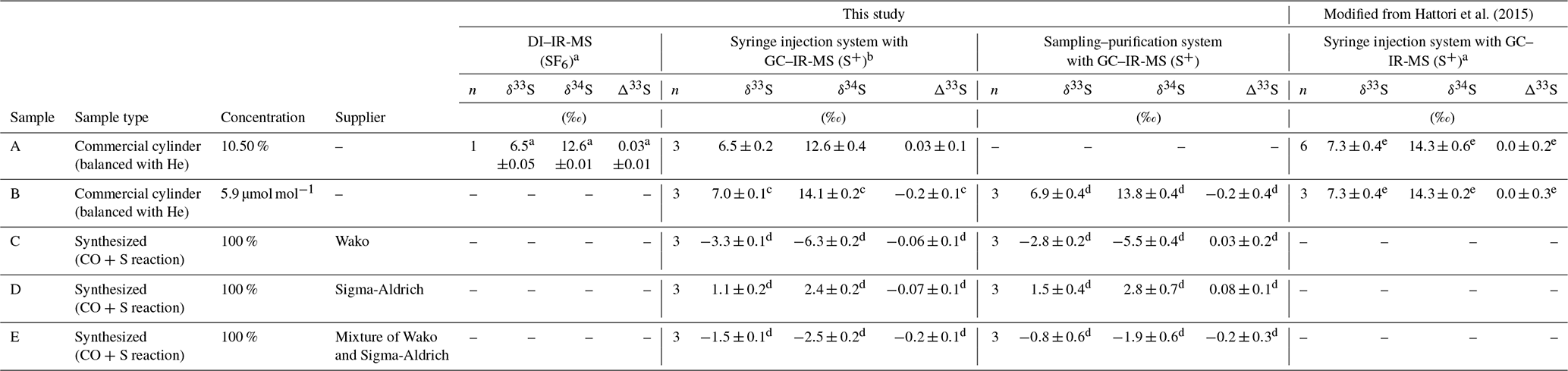
a δ33S, δ33S,
and Δ33S values of SF6 chemically converted from OCS in sample
A were corrected to values relative to the international standard (VCDT)
notation by SF6 converted from IAEA-S-1(Ag2S : δ33S =
−0.055 ‰, Ono et al., 2007; δ34S =
−0.30 ‰,
Robinson, 1993; and Δ33S =
−0.100 ‰, Ono et al., 2007), and the standard deviations (1σ)
of the measurements were estimated based on measurements repeated 25 times.
b System developed by Hattori et al. (2015).
c Corrected to values relative to the
international standard (VCDT) notation by using sample A measured in this study.
d Corrected to values relative to the
international standard (VCDT) notation by using daily sample B injected
from the line developed by Hattori et al. (2015).
e Average and precision of
δ34S(SF6) value chemically converted from OCS in sample A was
corrected to values relative to the international standard (VCDT) notation by
SF6 converted from IAEA-S-1(Ag2S : δ33S =
−0.055 ‰ Ono et al., 2007;
δ34S =
−0.30 ‰, Robinson, 1993; and Δ33S =
−0.100 ‰ Ono et al., 2007, Hattori et al., 2015).
For the testing of repeatability and collection efficiency of the systems, we used three commercially available cylinders of compressed air samples collected in Kawasaki, Japan (Toho Sanso Kogyo Co., Ltd., Yokohama, Japan), sample F (collected on 25 July 2017), sample G (collected on 2 July 2012), sample H (collected on 2 December 2017), sample I (collected on 26 October 2018), sample J (collected on 1 December 2018), and sample K (collected on 26 December 2018) (Table 2). These compressed air samples in these cylinders are collected with a compressor (YS85-V; Toa Diving Apparatus Co., Ltd., Tokyo, Japan) and are not dried. Sample G was used as sample E for Hattori et al. (2015). Its δ34S(OCS) value was (4.9±0.3) ‰. It was postulated as the global representative value at that moment. All compressed air cylinders are made of manganese steel without special wall treatments, engendering concerns about OCS decomposition in the cylinders.
Table 2Sample information for compressed air in cylinders collected at Kawasaki, Japan.

a Measured using a Q-MS with a picomole-level calibration curve. b Measured using a Q-MS with a nanomole-level calibration curve after sampling. c Measured using an IR-MS with a calibration curve in Fig. 4 after sampling with 1σ uncertainty of 6 %. d The previous δ34S(OCS) value measured by Hattori et al. (2015) was (4.9±0.3) ‰.
2.2 Sampling system
A schematic diagram of the sampling system is depicted in Fig. 1. The sampling system size and weight are 50 cm × 50 cm × 50 cm (width × height × depth) and 4 kg, except for a dewar (37 cm outer diameter, 66 cm height, and 11 kg weight) (MVE SC 20∕20; Chart Industries Inc., Georgia, USA). For field campaigns, the system can be easily disassembled and transported in two containers of 40 cm × 30 cm × 20 cm (width × height × depth). Reassembling the sampling system on site can easily be done within 2 h, making it suitable for field campaigns. The main compartments of the sampling system are 1∕4 in. (0.64 cm) PTFE tubes, 1∕8 in. (0.32 cm) stainless steel tubes, 1∕16 in. (0.16 cm) Sulfinert-treated stainless steel tubes (Restek Corp., PA, USA), Sulfinert-treated stainless steel ball valves V1, V2, V5, and V9, and stainless steel ball valves V3, V4, V6, V7, and V8 behind the sampling tube (Fig. 1). Excluding union tees made of stainless steel immediately before the pump, union tees coming in contact with the sampled OCS are made of Sulfinert-treated stainless steel (Fig. 1).
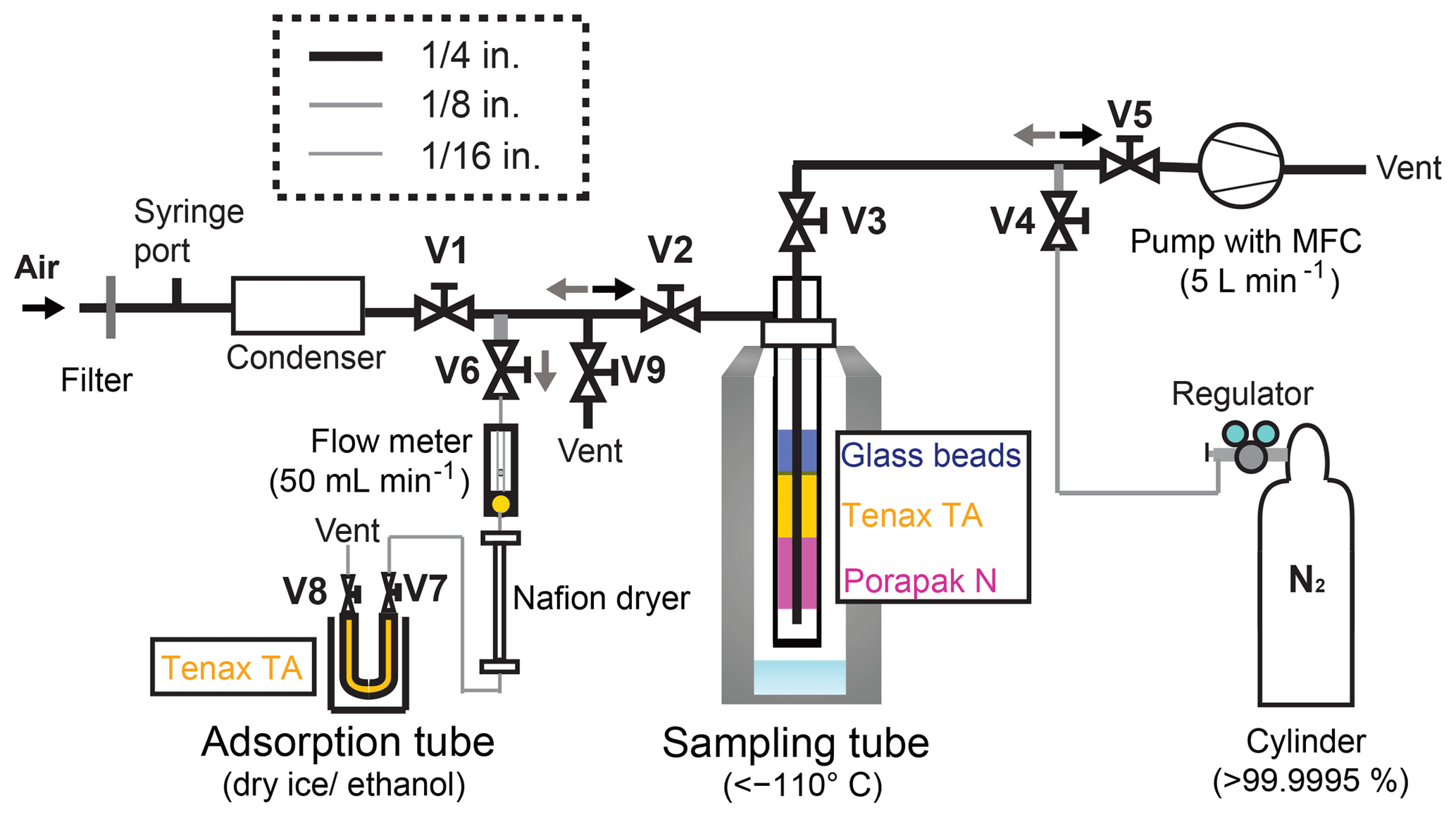
Figure 1Schematic diagram of the OCS sampling system. System components: V, valve; pump, vacuum pump; MFC, mass flow controller.
The cryotrap sampling tube for OCS concentration from ambient air consists of an outer stainless steel tube (3∕4 in. (1.9 cm) outer diameter, 50 cm length) with an air inlet at the side 4 cm below the top and an inner 1∕4 in. (0.63 cm) stainless steel tube (Bahlmann et al., 2011). From top to bottom, the sampling tube package is the following: 0–30 cm, empty; 30–40 cm, silanized glass beads 2 mm; 40–43 cm, Tenax TA (60∕80 mesh; GL Sciences Inc., Tokyo, Japan); 43–47 cm, Porapak N (80∕100 mesh; Sigma-Aldrich Corp., Japan); 47–50 cm, empty, and adsorbents separated by plugs of precleaned glass wools (GL Sciences Inc., Tokyo, Japan). We developed this sampling tube according to Bahlmann et al. (2011). Detailed functions of the respective components are described therein. Briefly, the glass bead traps the remaining water vapour from the sampled air and prevents water vapour adsorption on the Tenax TA and Porapak N. The glass bead further increases the temperature exchange between the cryotrap walls and the sampled air. The Tenax TA and Porapak N can be used for trapping volatile organic compounds. We assume that OCS is sampled on the Tenax TA and Porapak N, but most OCS might be trapped on Tenax TA. Although some components might not be necessary for OCS collections, up to this point, it has been working well for OCS sampling.
The adsorption tube consists of a stainless steel tube (with 1∕4 in. (0.63 cm) outer diameter, 17.5 cm length) filled with Tenax TA. Before experiments, the sampling tube and the adsorption tube were conditioned in the laboratory using 100 mL min−1 high-purity He flow at 160 ∘C with an electric heating mantle (P-22; Tokyo Technological Labo Co., Ltd., Kanagawa, Japan) for 6 h and 50 mL min−1 high-purity He flow at 330 ∘C with an electric heating mantle (P-25; Tokyo Technological Labo Co., Ltd.) for 6 h, respectively. We confirmed that possible contamination of OCS in the tubes was less than 10 pmol after conditioning. We also confirmed that the surface was inert for at least 3 days and that the inactive state of the surface of adsorbents in these tubes would be maintained under a no-leakage condition. It is noteworthy that conditioning steps would be required if stainless tubes are replaced by Sulfinert-treated tubes/valves because this conditioning was aimed at removing strongly adsorbed volatile organic compounds such as ethanol and acetaldehyde in adsorbents, which might interfere with OCS collection and/or react with OCS.
During sampling, valves V1, V2, V3, and V4 were opened. Then atmospheric samples were drawn with a low-volume diaphragm pump (LV-40BW; Sibata Scientific Technology Ltd., Saitama, Japan) through the sampling system with a flow of (5.00±0.25) L min−1. The air was first passed through a membrane filter (47 mm diameter, 1.2 µm pore, Pall Ultipor N66 sterilizing-grade filter; Pall Corp., New York, USA) set in a NILU filter holder system (70 mm diameter, 90 mm length: Tokyo Dylec Corp., Tokyo, Japan) to remove atmospheric aerosol. Then it was directed through a condenser (EFG5-10; IAC Co. Ltd., Japan) kept at approximately −15 ∘C to remove water vapour from the air. The air was then passed through the sampling tube at temperatures of −140 to −110 ∘C by vapour of the liquid N2 in a dewar. The OCS was enriched in the sampling tube, whereas other main gases (N2, O2, Ar, etc.) were passed through the sampling tube.
After sampling, valves V1 and V4 were closed, and valves V5, V6, V7, and V8 were opened. Then, the sampling tube was removed carefully from the dewar manually and was heated gradually to 130 ∘C. The vaporised gases in the sampling tube were passed to the adsorption tube cooled at −78 ∘C using dry ice–ethanol after removal of the remaining water vapour by a Nafion dryer (MD-110-96S; Perma Pure LLC, NJ, USA). The flow rate was regulated (approx. 50 mL min−1) by a needle valve equipped with a flow meter for 20 min. After the flow rate became lower than 10 mL min−1, V4 was opened. The sampling tube was flushed with pure N2 (> 99.99995 vol. %) at 50 mL min−1 for 40 min. After the transfer of samples, V6, V7, and V8 were closed. Then OCS was preserved in the adsorption tube. We initially confirmed that OCS did not pass through an adsorption tube at a flow rate lower than 50 mL min−1 using two adsorption tubes connected in series from the second adsorption tube: OCS was observed only from the first tube, not from the second tube. For this study, the collected OCS samples in adsorption tubes were measured within 30 min, except for the preservation test.
2.3 Purification system
After sampling OCS from the air using the sampling system as described above, the collected OCS was purified and connected directly to the measurement system. The schematic system is shown in Fig. 2. Excluding a fused silica capillary tube, all tubes and valves are made of stainless steel. U-shaped trap 1 is a 50 cm, 1∕4 in. (0.64 cm) outer diameter (1∕8 in. (0.32 cm) inner diameter) stainless steel tube. U-shaped trap 2 is a 30 cm, 1∕8 in. (0.32 cm) outer diameter (1∕16 in. (0.16 cm) inner diameter) stainless steel tube filled with Tenax TA (60∕80 mesh; GL Sciences Inc.). Before the experiment, trap 2 is heated to 150 ∘C by an electric heating mantle (P-22; Tokyo Technological Labo Co., Ltd.) for 30 min at 30 mL min−1 with high-purity He for conditioning. Coil-shaped trap 3 is an empty stainless steel tube (1∕16 in. (0.16 cm) outer diameter, 50 cm length). Coil-shaped trap 4 is a fused silica capillary tube (0.32 mm inner diameter, 50 cm length, GL Sciences Inc.). The GC1 (GC-8610T; JEOL Ltd., Tokyo, Japan) is equipped with a column packed with Porapak Q (80∕100, GL Sciences Inc.) (1∕8 in. (0.32 cm) outer diameter, 3 m length) to separate OCS from CO2. The GC1 oven temperature for OCS purification was programmed to provide 30 ∘C for 5 min, increasing to 60 ∘C at 30 ∘C min−1, followed by an increase to 230 ∘C at 30 ∘C min−1 starting 40 min after the start of the program for GC1, and 230 ∘C for 1 min.
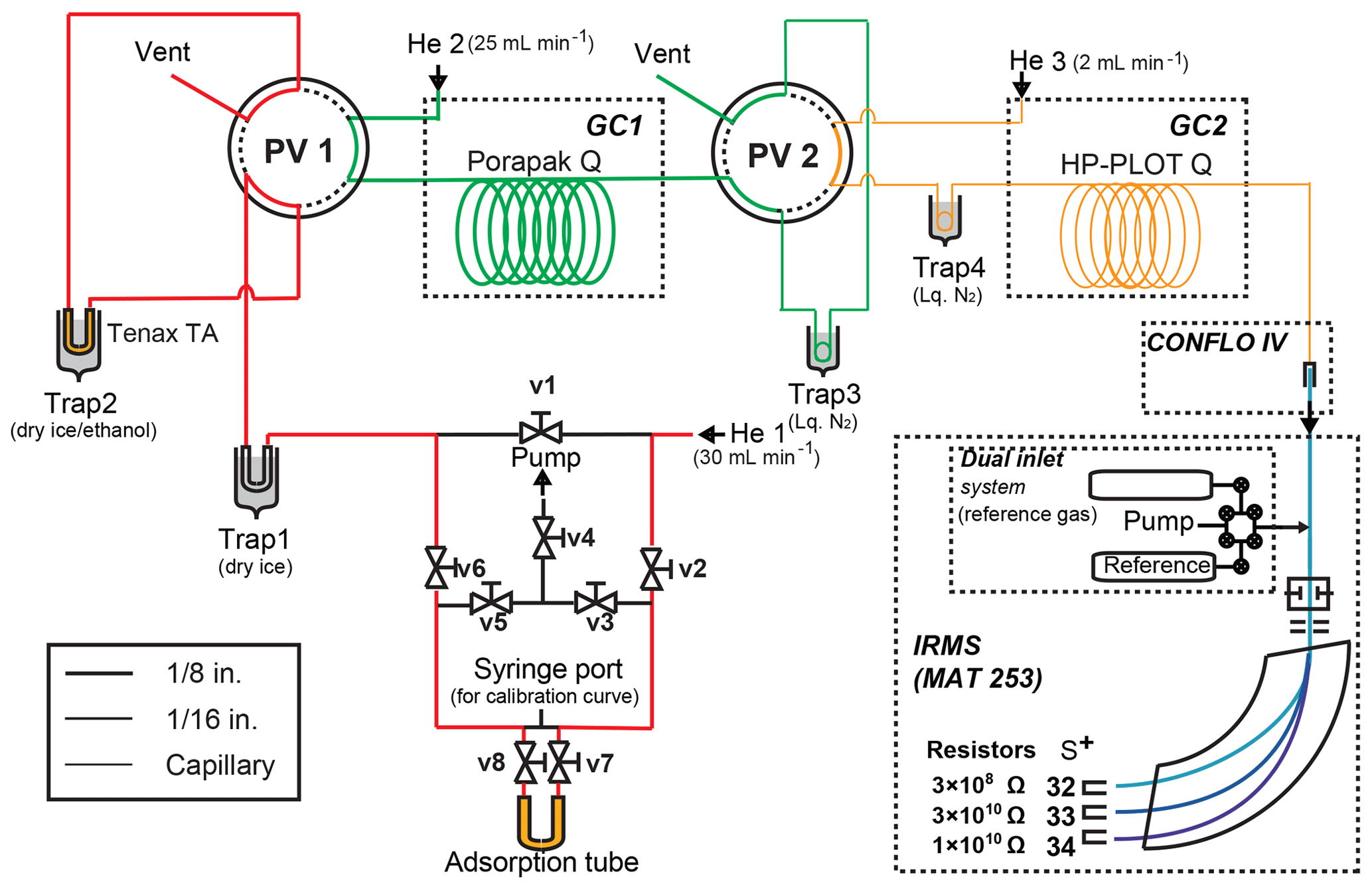
Figure 2Schematic diagram of the OCS purification system. System components: V, valve; pump, vacuum pump; MFC, mass flow controller.
After the adsorption tube containing OCS was connected to the purification system, v3, v4, and v5 (Fig. 2) were opened and the air in the line was pumped out using a rotary pump (DA-60D; Ulvac Kiko, Miyazaki, Japan) for 5 min; v3, v4, and v5 (Fig. 2) were then closed. When the adsorption tube was heated at 130 ∘C and v2, v7, v8, and v6 (Fig. 2) were opened, gases in the adsorption tube passed through trap 1 cooled by dry ice (−78 ∘C) to remove trace remnant water vapour. Also, OCS was collected in trap 2, with Tenax TA cooled by dry ice–ethanol (−72 ∘C) with a high-purity He flow rate of 30 mL min−1. After 15 min, port valve (PV) 1 was changed. Trap 2 was then removed from dry ice–ethanol and was heated at 130 ∘C. The retention times of CO2 and OCS were initially determined by injecting a mixture of 8 mmol of CO2 from pure CO2 in a cylinder (99.995 % purity; Japan Fine Products Co. Ltd.) and 10 nmol of OCS from sample C. They were 3–10 min for CO2 and 20–30 min for OCS at a flow rate of 25 mL min−1. Trap 3 was cooled by liquid N2 starting 10 min after the start of the program for GC1; PV2 was changed from 15 to 35 min after injection of samples to GC1 to introduce OCS to trap 3. OCS with high-purity He was passed through the column and collected in trap 3 for 20 min. After OCS collection in trap 3, the OCS was again transferred to trap 4 in liquid N2 at 6 mL min−1 by high-purity He with removal of liquid N2 from trap 3 to a cryofocus. Trap 4 was then removed from liquid N2; the OCS passed through the GC2 and was introduced directly to the detectors (quadrupole mass spectrometer (Q-MS) or IR-MS depending on the experiments explained below).
2.4 Determination of the OCS concentration
The OCS concentrations were measured with a GC–Q-MS (7890A; Agilent Technologies Inc., CA, US, coupled to Q-MS, 5975C; Agilent Technologies Inc., CA, USA) equipped with a capillary column (0.32 mm inner diameter, 25 m length, and 10 µm thickness; HP-PLOT Q, Agilent Technologies, CA, USA). The He flow was set to 1.5 mL min−1 and the oven temperature program was set as 60 ∘C for 15 min, increased to 230 ∘C at 60 ∘C min−1, and then held at 230 ∘C for 1 min.
To ascertain the OCS concentration of sample A once a month, and to ascertain the collected OCS amounts using a sampling system, we made a calibration curve for OCS ranging from 0.1 nmol to 10 nmol using a Q-MS. The calibration curve for the nanomole level is calculated with an injection of sample B with a volume of 0.5, 2.2, 4.4, 8.8, 11, 13.2, 17.6, 22, and 44 mL (n=3). The precision (standard deviation (1σ) relative to mean) of the OCS amount from a syringe injection was estimated to be ±3 % by the standard deviation of the relative error between the measured values and the estimated value for calibration curves.
To ascertain the OCS concentrations of samples F and G, we prepared calibration curves for OCS ranging from 0 to 100 pmol using a Q-MS. The calibration curve for the picomole level is calculated from an injection of sample B with a volume of 0, 200, 400, and 800 µL (n=3) with a precision of ±3 % as estimated similarly above. For determination of OCS concentrations of samples F and G, samples F and G were stored in 50 mL two-neck glass bottles with atmospheric pressure and were introduced to the purification system from an attached glass bottle instead of an adsorption tube. The measured OCS concentrations for samples F and G were, respectively, (380 ± 15) pmol mol−1 and (160 ± 5) pmol mol−1 (Table 2).
The OCS concentrations for samples F, G, H, and I were lower than typical atmospheric OCS concentrations (400–550 pmol mol−1) (Montzka et al., 2007), even though the samples were compressed air collected from the ambient atmosphere. Because we were concerned about the changes in OCS concentrations for samples F and G, the OCS concentrations for samples F and G were measured within at least a week before or after the experiment. In a similar manner, the cylinders of samples H, I, J, and K were used for experiments within 2–3 days. Therefore, a change in OCS concentration in samples might occur.
2.5 Determination of the sulfur isotope ratios of OCS
For determination of the sulfur isotope ratios of OCS, OCS was passed through the GC2 after a purification system as described above. Then it was introduced directly to the IR-MS (MAT253; Thermo Fisher Scientific Inc., Berlin, Germany) via an open split interface (ConFlo IV; Thermo Fisher Scientific Inc.). Reference OCS of sample A was purified with liquid N2 (−196 ∘C) and then introduced via a conventional dual inlet system. Pure OCS is not commercially available in Japan because of its toxicity (Hattori et al., 2015). In addition to the method introducing OCS to the IR-MS as described above, the conventional syringe injection line, which was previously used for Hattori et al. (2015) and Kamezaki et al. (2016), was also used for comparison or calibration. Briefly, the syringe-injected OCS was collected in stainless steel tubes (10.5 mm inner diameter, 150 mm length) cooled at −196 ∘C by liquid N2 with a gentle vacuum by a rotary pump (Pascal, 2010; Pfeiffer Vacuum GmbH, Aßlar, Germany) with regulation using a valve. After transfer of OCS to the trap, the two-way six-port valve was changed. Then liquid N2 was removed from the trap. Subsequently, OCS was transferred and collected in a fused silica capillary tube (0.32 mm inner diameter, 50 cm length; GL Sciences Inc.) covered by a stainless steel tube containing liquid N2 for 13 min before being introduced into the GC–IR-MS system.
In the IR-MS ion source, electron impact ionization of OCS produced S+ fragment ions. The sulfur isotope ratios in OCS were therefore determined by measuring the fragment ions 32S+, 33S+, and 34S+ using triple Faraday collector cups. The typical precisions (1σ) of the replicate measurements (n=3) are 0.4 ‰, 0.2 ‰, and 0.3 ‰ for δ33S(OCS), δ34S(OCS), and Δ33S(OCS) values, respectively. A reference OCS gas was introduced for 20 s three times starting at t=350, 825, and 1025 s. The reference gas at t=350 s was used as the reference for all calculations of OCS sulfur isotope ratios. To remove hydrogen sulfide and ethane from ambient samples, from t=300 s, the effluent from the GC column was kept off the MS line using back-flushed helium flow. Sulfur isotope ratios are typically reported as
where xR represents the isotopic ratios (xS∕32S, where x=33 or 34) of the samples and standards. The sulfur isotope ratios are reported relative to the Vienna Canyon Diablo Troilite (VCDT, quoted as per mil values (‰)). In addition to the δ values, capital delta notation (Δ33S value) is used to distinguish mass-independent fractionation (MIF; or non-mass-dependent fractionation) of sulfur, which causes deviation from the mass-dependent fractionation (MDF) line. The Δ33S value describes the excess or deficiency of 33S relative to a reference MDF line. It is expressed as
The δ values in this study were determined using the following processes. First, we ascertained the δ34S value of sample A by converting OCS to SF6. The δ34S(SF6) value was measured relative to the VCDT scale by comparing SF6 similarly converted from IAEA-S-1 (Ag2S: ‰; Robinson, 1993) as described by Hattori et al. (2015). The measured δ34S value of sample A was 12.6 ‰, which was lower than the data presented by Hattori et al. (2015) with 14.3 ‰ (Table 1). Secondly, the δ34S value of sample B, which was used as a working standard for δ34S measurements, was ascertained from comparison with the δ34S value (at VCDT scale) of sample A with the GC–IR-MS method using a S+ fragment ion. The δ34S(OCS) value of sample B in this study was (14.1±0.2) ‰ (Table 1), showing no significant difference with the δ34S(OCS) value of sample B (14.3±0.2) ‰ in data presented by Hattori et al. (2015). It is noteworthy that we also found that the OCS concentration in sample B was not changed. Sample B was used as the daily working standard for GC–IR-MS measurement to ascertain sample δ34S(OCS) values for other samples used throughout this study (Table 1).
3.1 Sampling efficiency of OCS
To test the sampling and desorption efficiency, the cylinder containing sample F was connected to a flow meter and the flow was adjusted to 6 L min−1 with a needle valve. An amount of 5 L min−1 was drawn through the sampling system with a pump and the remainder was vented into the air to maintain atmospheric pressure at the sampling inlet. The samples were collected within 2 days to prevent OCS loss in the cylinder. The vent flow was measured with a flow meter (ACM-1A; Kofloc, Tokyo, Japan). To ascertain the trapping efficiency OCS was sampled for 10, 50, and 100 min with blank test intervals as presented in Fig. 3a (see Sect. 2.2 for sampling procedure). The sampling times corresponded to sampling volumes of (50±2.5) L, (250±13) L, and (500±25) L and the corresponding OCS amounts were (0.77±0.04) nmol, (3.9±0.2) nmol, and (7.7±0.4) nmol respectively (Fig. 3a).
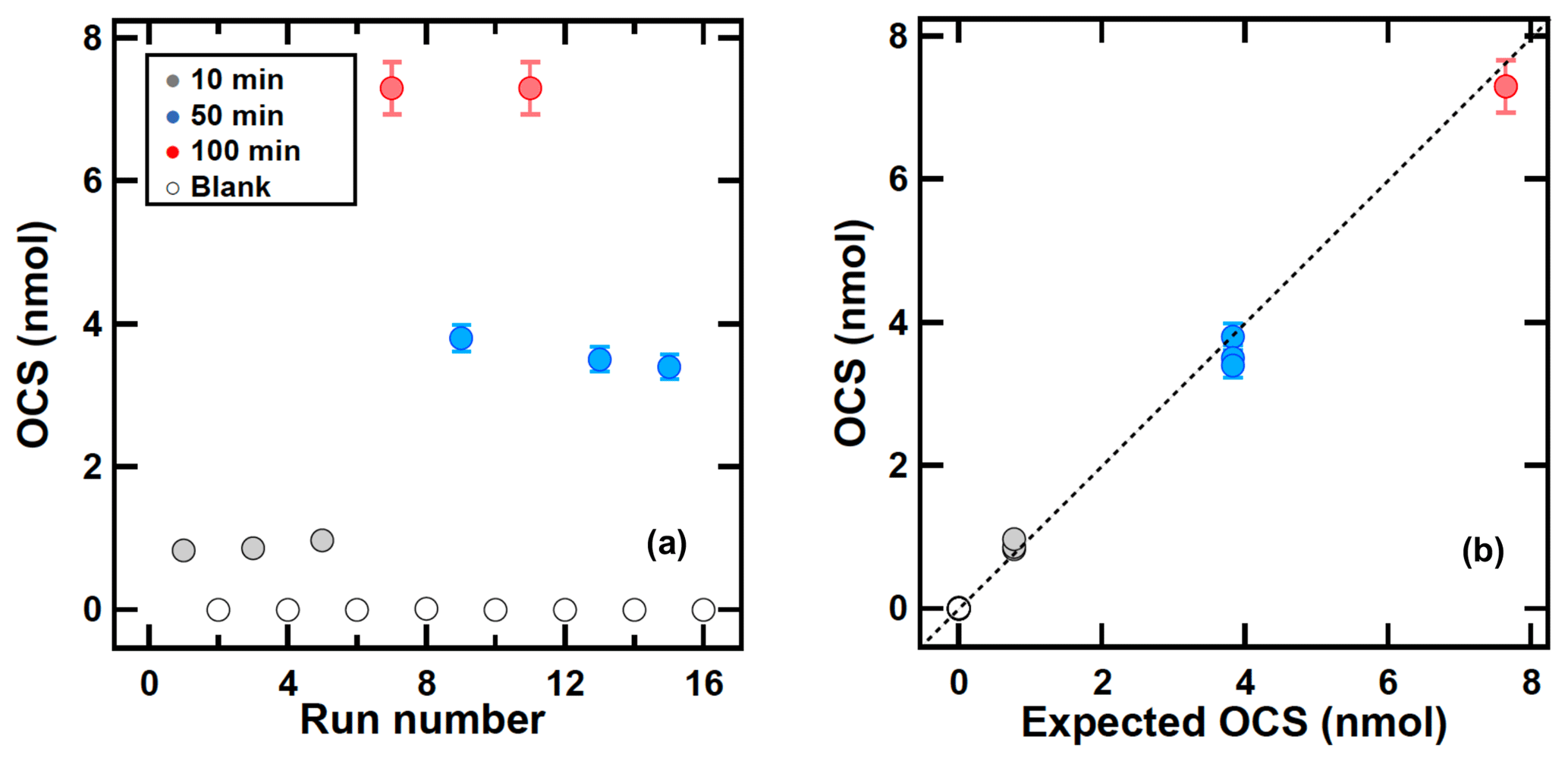
Figure 3OCS sampling using sample F of (380±15) pmol mol−1 with different sampling times of blank (0 min), 10, 50, and 100 min. (a) Collected OCS amounts as a function of run numbers. (b) Observed OCS amounts and OCS amounts calculated using OCS concentration multiplied by the sampling time. The error bar shows ±3 % based on the residual of measured OCS peak area and calibrated OCS peak area. The dotted line shows the slope of x=y.
Recovery and precision (1σ) for OCS amounts collected for sampling times of 10, 50, and 100 min were (0.9±0.1) nmol (n=3), (3.6±0.2) nmol (n=3), and (7.4±0.3) nmol (n=2), respectively. The OCS blanks were smaller than 15 pmol. These results indicate that the yield of OCS during sampling and transferring from the sampling tube to the adsorption tube is almost over 95 %. The memory effect of the system between the sampling runs is expected to be less than 1 % when sampling OCS amounts over 3 nmol (approx. 50 min). Figure 3b presents a comparison of OCS amount between observed OCS amounts and OCS amounts calculated based on OCS concentration in sample F and sampling time, showing that all results fall on the 1:1 line. This suggests that almost 100 % of OCS for sampling runs was collected in the sampling tube and was transferred successfully to the adsorption tube. Although the collected OCS amount in 10 min was slightly larger than the expected OCS amount, the OCS amounts in 100 min were slightly lower than the expected OCS amount. This result indicates that a small OCS contamination during the sampling and a purification system might exist but that it might not be significant, as discussed above.
3.2 Accuracy of the sulfur isotopic analysis of OCS via sampling–purification systems
In the developed system, the possibility exists that OCS is lost by passing OCS through GC1. Also, because the flow rate of approximately 50 mL min−1 was lower than the flow rate of approximately 200 mL min−1 reported by Hattori et al. (2015), the possibility exists that OCS was lost by trap 1. Therefore, to assess these possibilities, the following test was conducted. Firstly, 5 nmol of OCS was injected to a system consisting of trap 2, GC2, and trap 4 and measured as a true value. Then the same amount of OCS was introduced into the developed purification system and the amount of OCS obtained was compared to the true value. These tests revealed an OCS loss of less than 2 % using a newly developed method and suggest a complete recovery of OCS within the given limits of uncertainty (±3 %). To assess the dependence of the sulfur isotopic measurements on the OCS amount, different amounts of OCS using sample B were tested. We introduced aliquots of 3, 6, 10, and 15 nmol of sample B over 30 min with a gas-tight syringe via a syringe port made from a tee union with a septum. The syringe port was placed between the inlet filter and the condenser and the sampling inlet was connected to high-purity N2 (99.99995 vol. %; Nissan Tanaka Corp., Saitama, Japan) (Fig. 1). For each experiment, a total volume of 500 L N2 was processed. The OCS contamination for this experiment was (0.30±0.16) nmol (n=3) when we flushed with 500 L of pure N2. For comparison, similar amounts of OCS were also injected using a syringe injection system developed previously (Hattori et al., 2015). Comparisons of OCS concentrations and δ and Δ values are depicted in Fig. 4. Although the observed OCS isotope ratios using 3 nmol of OCS with the developed method were scattered (1σ uncertainty: 1.0 ‰, 1.0 ‰, and 0.5 ‰, respectively, for δ33S(OCS), δ34S(OCS), and Δ33S(OCS) values), the reproducibilities at the 6 nmol level were sufficient (1σ uncertainty: 0.4 ‰, 0.2 ‰, and 0.4 ‰, respectively, for δ33S(OCS), δ34S(OCS), and Δ33S(OCS) values) and were similar to those obtained with the conventional syringe injection system for Hattori et al. (2015) (Fig. 4). Consequently, this system better accommodates OCS samples over 6 nmol, indicating the necessity for collection of ambient air in amounts greater than 300 L.

Figure 4OCS amounts and sulfur isotope ratios of different amounts of OCS injections ascertained using the developed sampling–purification system and conventional syringe injection system (Hattori et al., 2015): (a) OCS amount; (b) δ33S; (c) δ34S; (d) Δ33S; closed symbols, sampling–purification system developed for this study; open symbols, conventional syringe injection system. All sulfur isotope ratios are relative to VCDT. The error bars are 1σ of the measurements based on triplicated measurements.
Furthermore, to test possible sulfur isotopic fractionations during sampling–purification processes, which might change the measurement accuracy, we compared the developed sampling–purification system with the conventional syringe injection system using 8 nmol of the in-house synthesized OCS (samples B, C, D, and E) with triplicate injections. In Fig. 5, the δ34S(OCS) values measured using the developed sampling–purification system were 0.2 ‰ lower (sample B) but 0.8 ‰, 0.4 ‰, and 0.6 ‰ higher (samples C, D, and E, respectively) than those measured using the syringe injection system of Hattori et al. (2015) (Table 1). This phenomenon was observed similarly for the δ33S(OCS) values (Fig. 5c), indicating that this process is not isotopic fractionation but rather suggests contamination during the sampling processes. When considering (0.30±0.16) nmol OCS (i.e. approx. 4 % for 8 nmol OCS samples) with δ34S of 3 ‰–18 ‰ covering the reported δ34S range of OCS sources (Newman et al., 1991), the accuracy of the δ34S(OCS) can be shifted from −0.3 ‰ to +0.3 ‰. Because the precision of 1σ uncertainty is 0.2 ‰, the overall precision values (1σ) for δ34S of this sampling–purification system were estimated as 0.4 ‰.
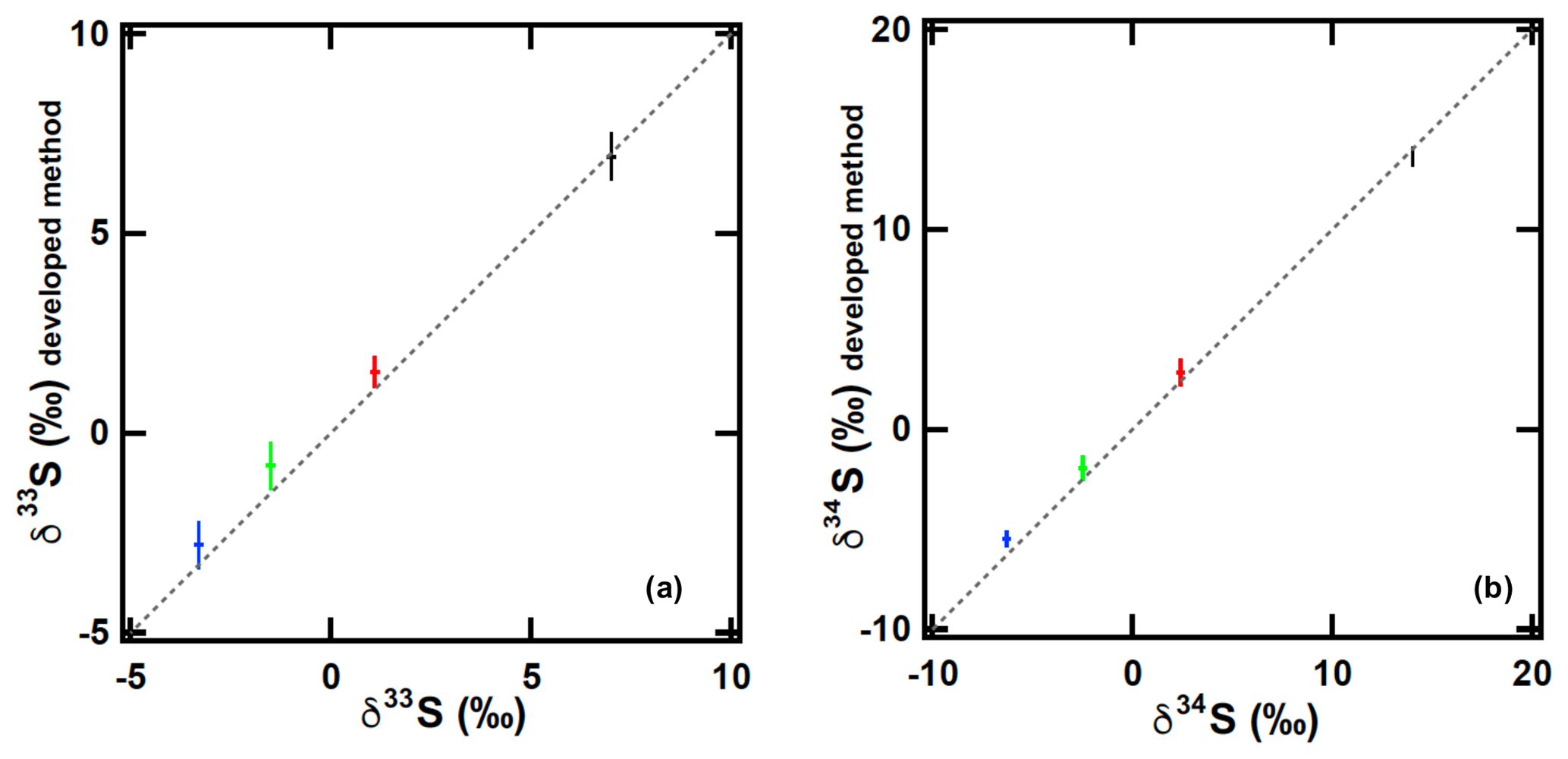
Figure 5Sulfur isotope ratios (a δ33S and b δ34S) ascertained from the developed sampling–purification system (y axis) and conventional syringe injection system (Hattori et al., 2015) (x axis). OCS sample amounts are 8 nmol. Different colours represent different samples: black, sample B; red, sample C; green, sample D; blue, sample E. The dotted line shows the slope x=y. The error bar is 1σ of each amount of triplicated OCS injection.
3.3 Sulfur isotope ratio for atmospheric OCS
Four ambient air samples were collected at the Suzukakedai campus of the Tokyo Institute of Technology located in Yokohama, Japan (35.5∘ N, 139.5∘ W, 27 m height), during 23–25 April 2018 every 12 h (sampling times were 23 April 2018 at 12:00 JST, 24 April 2018 at 00:00, 24 April 2018 at 12:00, and 25 April 2018 at 00:00). The sampling volume was 500 L (i.e. 100 min with a pump flow of 5 L min−1). Measurements of OCS concentrations and sulfur isotope ratios were carried out within 30 min after the sampling. The time for a single measurement of δ34S value for atmospheric OCS was 100 min (500 L) for sampling of air, 40 min for transferring to the adsorption tube, 40 min for purification, and 20 min for measurement using an IR-MS. The OCS concentrations and δ34S(OCS) values observed for ambient air are presented in Fig. 6.

Figure 6OCS concentrations and δ34S(OCS) values for atmospheric samples collected at the Suzukakedai campus of the Tokyo Institute of Technology located in Yokohama, Japan. The error bar is 6 % for OCS concentration based on the precisions of syringe injection and flow rate of the diaphragm pump in the sampling system. The precision of δ34S is estimated from a 1σ uncertainty of 0.4 ‰.
In contrast to the δ34S(OCS) value, the δ33S(OCS) value in air was not determined because of the unexpected peak (approx. 40 mV height) observed for m∕z 33, which slightly overlapped the OCS peak of the chromatogram (Fig. 7). We notably did not observe any interferences on m∕z 32 and m∕z 34. The interfering compound could have not yet been identified. Known fragments interfering on m∕z 33 are CH5O+ originating from the protonation of methanol and/or the reaction of CH3O+ with H2O, CH2F+ that is indicative of hydrofluorocarbons, and/or NF+ deriving from nitrogen trifluoride (NF3). To measure m∕z 33 of OCS without interferences, further improvement of peak separation of OCS with interferences is required by changing the parameter of the separation in the system and/or data processing. For example, a custom-made MATLAB routine, which can extrapolate the peak tail of interference via an exponentially decaying function to distinguish the two gaseous species as described in Zuiderweg et al. (2013), would enable us to analyse m∕z 33 in addition to the standard ISODAT software used for isotope ratio measurements.
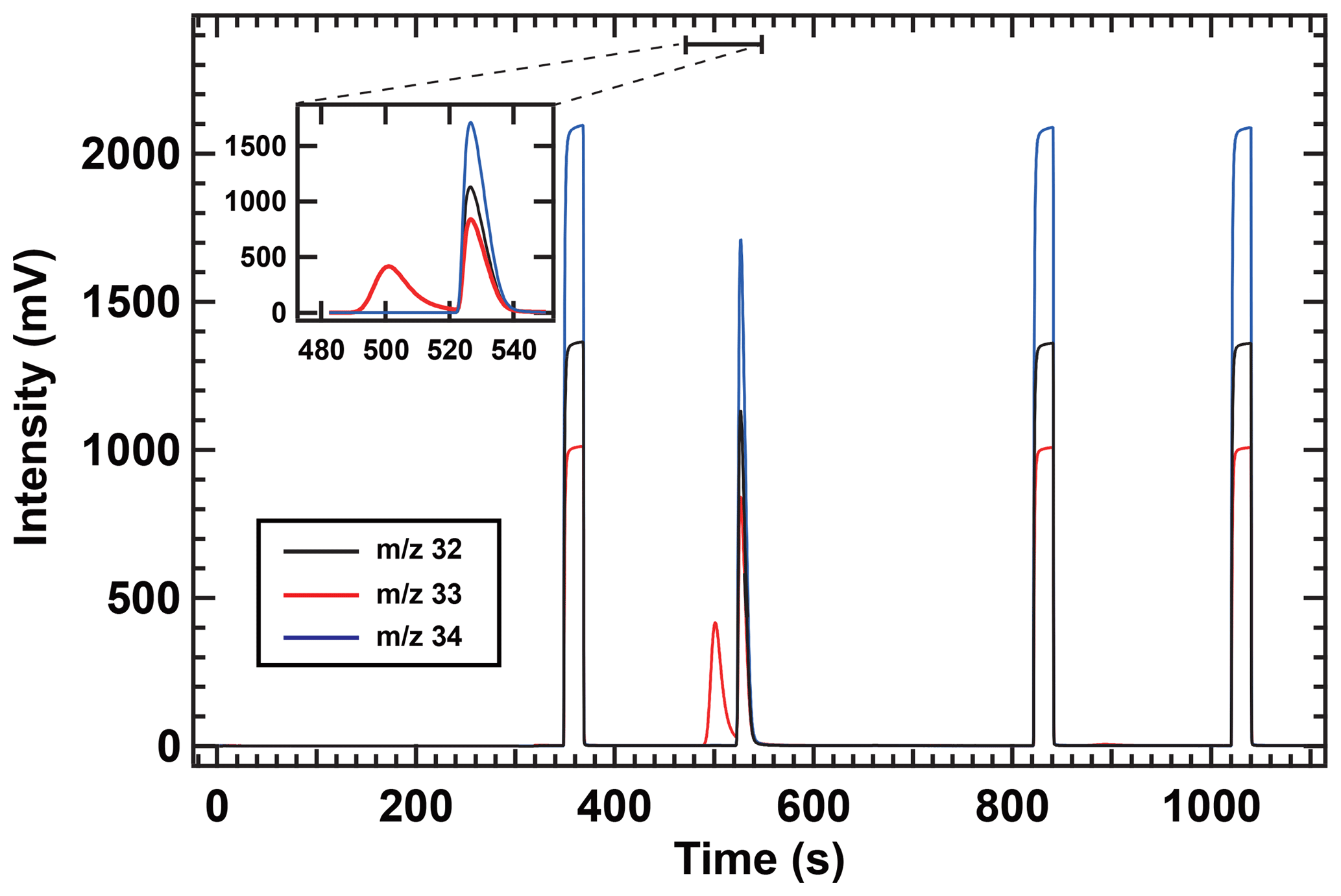
Figure 7IR-MS chromatogram of atmospheric samples collected at the Suzukakedai campus of the Tokyo Institute of Technology. Liquid N2 removal from trap 4 occurred at 0 s in the purification system. Reference OCS was injected three times starting at 350, 825, and 1025 s for 20 s.
The observed OCS concentrations for atmospheric samples were 447–520 pmol mol−1 (Fig. 6a), averaging (492±34) pmol mol−1. Data show no clear differences between 00:00 and 12:00 in 2 days (p value = 0.65). This OCS concentration observed at the Suzukakedai campus shows good agreement with the OCS concentrations observed at a similar latitude in the US (e.g. 400–550 pmol mol−1; Montzka et al., 2007). Berkelhammer et al. (2014) reported diurnal variation for OCS concentrations in the US with the lowest at 08:00 and the highest at 16:00 with 80 pmol mol−1 changes in a day. Moreover, the differences of OCS concentrations for four atmospheric samples were less than 80 pmol mol−1. The observed δ34S(OCS) values of four atmospheric samples were 10.4 ‰–10.7 ‰ (Fig. 6b) and averaged (10.5±0.4) ‰. The δ34S(OCS) values also showed no clear diurnal difference (p values = 0.29) (Fig. 6b). Given the diurnal OCS variations, some future study is clearly necessary to ascertain whether or not δ34S(OCS) values have diurnal variations by comparing δ34S(OCS) values for the highest OCS concentration at 08:00 and the lowest OCS concentration at 16:00.
It is noteworthy that the δ34S(OCS) values of four atmospheric samples were clearly distinct from our earlier observed δ34S(OCS) value of (4.9±0.3) ‰ obtained from sample G (Hattori et al., 2015), which was postulated as a global representative δ34S(OCS) value in the atmosphere. In fact, the OCS concentrations in the commercial cylinders F, G, and H were significantly lower than typical atmospheric OCS concentrations of approximately 500 nmol mol−1 (Table 2). Ascertaining the δ34S(OCS) value in sample G using the current sampling–purification system yielded a δ34S(OCS) value of (6.1±0.2) ‰ slightly higher than the previous value of (4.9±0.3) ‰ (Hattori et al., 2015). It is possible to explain this 1.2 ‰ increase for the δ34S(OCS) value for a case in which the contaminated OCS has a δ34S(OCS) value of over 17 ‰. However, such a high δ34S(OCS) value from contamination requires a situation in which the contaminated OCS comes only from the ocean, which is not likely. Because the atmospheric δ34S(OCS) values in this study were (10.5±0.4) ‰ and higher than that for sample G, the increased δ34S(OCS) values are expected to be affected by isotopic fractionation during OCS degradation in the cylinder and not by contamination. The causes for the OCS losses in the commercial pressurized air cylinders could not be investigated here. Indeed, as reported by Kamezaki et al. (2016), OCS is decomposed by hydrolysis, which increases the δ34S(OCS) value. Additionally, observation of OCS loss caused by adsorption to walls in the canister was reported by Khan et al. (2012). The compressed air of samples F and G might be affected by anthropogenic OCS sources at the sampling site and/or during the compressing processes. All in all, the δ34S(OCS) value of sample G is no longer considered to be a representative of atmospheric OCS.
3.4 Preservation tests
As described above, we measured OCS concentration and sulfur isotope ratio of atmospheric samples within 30 min after sampling. The OCS concentrations are consistent with the observed OCS concentrations in the same latitude and our tests revealed no OCS losses under these conditions. However, after the development of the system, we realized up to 50 % of OCS can be decomposed during storage of the adsorption tube after we have measured the samples within 14 days after sampling (Fig. 8a). We also found that the OCS in the stainless steel adsorption tubes stored at 25 ∘C showed only slight changes in concentration with () % and (0.2±0.4) ‰ for δ34S(OCS) values after 3 h (Fig. 8b). All data sets presented up to this point were undertaken immediately after the sampling (i.e. shorter than 30 min). Therefore, we did not expect marked changes in OCS concentrations and the δ34S(OCS) values for most datasets including atmospheric OCS samples. Because OCS is known to react with the surface of stainless steel (Khan et al., 2012), for future use this fact requires appropriate ways of preservation of OCS during transportation from field sampling sites to laboratory until analysis.
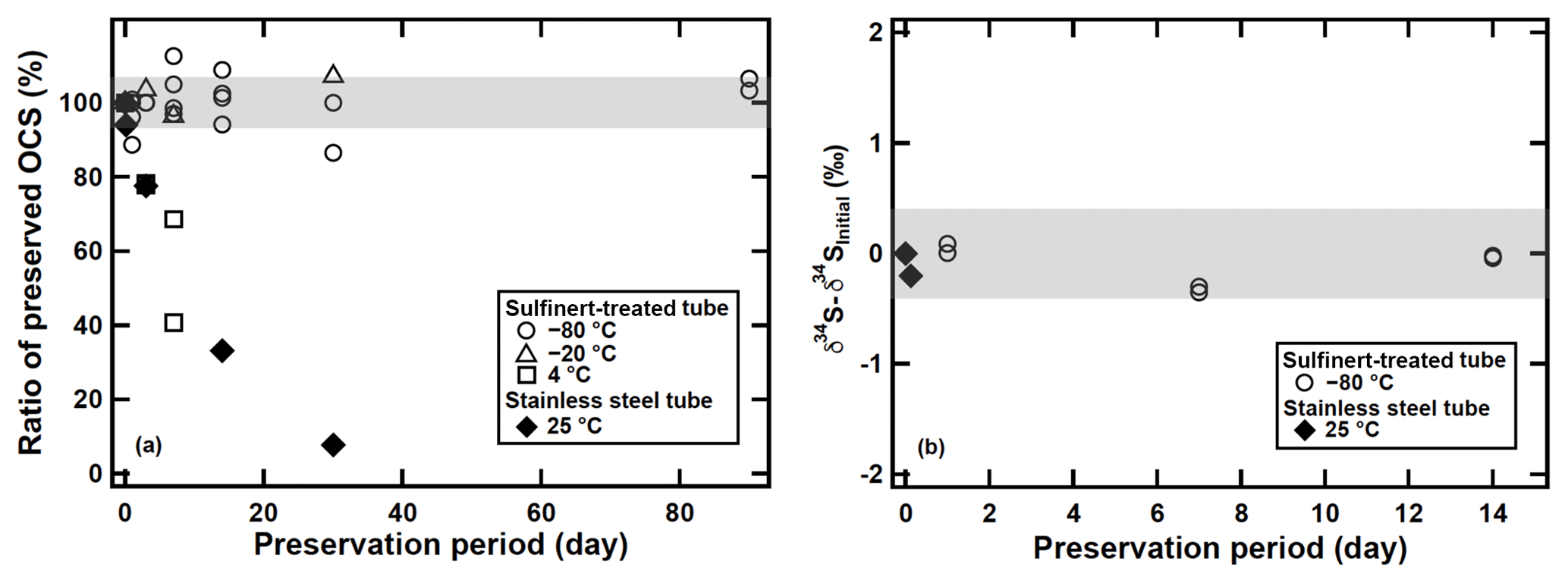
Figure 8(a) Changes in OCS concentrations preserved in the OCS storage test in adsorption tubes at different temperatures and tubes. (b) Changes in δ34S(OCS) preserved in the OCS storage test. The shaded bar shows ±6 % for OCS concentration and ±0.4 ‰ for the δ34S(OCS) value based on the precisions of syringe injection and the flow rate of the diaphragm pump in the sampling system.
In order to minimize potential OCS decomposition on the surface wall, we modified the adsorption tube by replacing the stainless steel tube and valves with a Sulfinert-treated tube and Sulfinert-treated valves. The preservation of OCS on the modified adsorption tubes at different storage temperatures was investigated using samples H, I, J, and K. The samples were processed as that described in Sect. 2.2 and transferred to the adsorption tubes. The adsorption tubes were stored at temperatures of 25, 4, −20, and −80 ∘C until measurements. After each storage period, the samples were analysed for OCS yields and δ34S(OCS) values as described in Sect. 2.3, 2.4, and 2.5. A rapid OCS decomposition of approximately 20 % during 7 days of storage was observed for the stainless steel adsorption tubes stored at 25 ∘C. A similar pronounced loss was observed for the Sulfinert-treated adsorption tubes stored at 4 ∘C but at a storage temperature of −20 ∘C. The OCS was stable for 30 days at −20 ∘C and for at least 90 days at −80 ∘C within a 1σ uncertainty of 6 % (Fig. 8a). Furthermore, we found that the δ34S(OCS) values showed no significant change during storage for at least 14 days at −80 ∘C (Fig. 8b). These results demonstrate that it is possible to apply this method for field campaigns by storing the adsorption tube at −80 ∘C after sampling.
3.5 Atmospheric implications
The δ34S(OCS) value of (10.5±0.4) ‰ is generally consistent with earlier estimation by Newman et al. (1991), who expected the mean δ34S(OCS) values of 11 ‰ based on the flux of continental emission to be 3 ‰ and oceanic emission to be 18 ‰ (Newman et al., 1991). This estimation is based on older information, but current measurements of atmospheric dimethyl sulfide (DMS) and dimethylsulfonioproionate (DMSP) are similar to 18 ‰ (Said-Ahmad and Amrani, 2013; Amrani et al., 2013; Oduro et al., 2012); continental sulfur sources also show approximately 0 ‰–5 ‰ (Tcherkez and Tea, 2013).
It is noteworthy that the potential importance of tropospheric sulfur isotopic fractionations during OCS sinks. To date, sulfur isotopic fractionations were reported as −5 ‰ to 0 ‰ for reaction with OH radical (Schmidt et al., 2012) and −2 ‰ to −4 ‰ for decomposition by soil microorganisms (Kamezaki et al., 2016; Ogawa et al., 2017). Sulfur isotopic fractionation for OCS by plant uptake, the dominant OCS sink in the troposphere (Berry et al., 2013), has not been determined, but the theoretical isotopic fractionation constant by plant uptake is −5.3 ‰ (Angert et al., 2019). Therefore, all sulfur isotopic fractionation constants by OCS degradation are negative, indicating that the δ34S(OCS) values can be increased by OCS degradation in the troposphere. Because the main OCS sink is photosynthesized by plants, the δ34S(OCS) values in the atmosphere might be increased in the growing season for plants in April. However, because of the long lifetime of OCS, δ34S(OCS) values might not be sensitive to seasonal variation. Future studies must be conducted to determine the isotopic fractionation constants and observations of δ34S(OCS) values to estimate the dynamics of atmospheric δ34S(OCS) values in the troposphere.
In addition to our observation of atmospheric δ34S(OCS) values with (10.5±0.4) ‰ at the Suzukakedai campus, Yokohama, Japan, δ34S(OCS) values of (13.4±0.5) ‰ in August–October at Israel, (12.8±0.5) ‰ in February–March in Israel, and (13.1±0.7) ‰ in February–March at the Canary Islands, Spain, were recently reported using the GC–MC–ICP-MS method (Angert et al., 2019). These differences indicate that the atmospheric δ34S(OCS) values might not be homogeneous, instead reflecting some geographic effects and/or potential difference for isotopic fractionations during sink processes. Given the higher influences of sulfur isotopic fractionations on δ34S(OCS) values during growing seasons, it is not likely to explain lower atmospheric δ34S(OCS) values for the Suzukakedai campus in April compared to those for Israel and the Canary Islands observed in February–March. Rather, δ34S(OCS) values of (10.5±0.4) ‰ at the Suzukakedai campus might be more affected by anthropogenic OCS emission and/or less affected by oceanic OCS emissions compared to the samples collected in Israel or the Canary Islands with higher δ34S(OCS) values. Potential anthropogenic OCS sources are Chinese emissions from rayon production (rayon yarn and staple rayon) and coal (industry and residential emissions), as pointed out by recent OCS source inventories (Zumkehr et al., 2018). In fact, the OCS concentration in the vicinity of China is high based on satellite observation (Glatthor et al., 2015). Future study is necessary to observe spatial and temporal variation of δ34S(OCS) values to discuss the link between anthropogenic activity and OCS cycles.
In addition to tropospheric OCS sources, OCS has some potential as a tracer of net ecosystem exchange into GPP on land (Campbell et al., 2008). Based on our earlier experiments, to elucidate OCS in the troposphere and its relation to biochemical activity by plant and soil microorganisms, OCS sulfur isotope analysis provides a new tool to investigate soil OCS sinks in the troposphere. To date, we have determined the isotopic fractionation constants for OCS undergoing bacterial OCS degradation and its enzyme (Kamezaki et al., 2016; Ogawa et al., 2017). Similarly, additional studies that include specific examination of isotopic fractionation by plant uptake, another major sink of atmospheric OCS, are indispensable for distinguishing the respective OCS fluxes of soil and plants. By coupling isotopic fractionations by soil and plant with atmospheric observations of δ34S(OCS) values using our newly developed method, the atmospheric observations of δ34S(OCS) values are expected to help refine estimates of biological activities of plant and soil microorganisms and their respective contributions to OCS degradation in the troposphere.
3.6 Comparison with other methods
Here we discuss the comparison of this sampling system coupled with the GC–IR-MS and GC–MC–ICP-MS methods (Said-Ahmad et al., 2017; Angert et al., 2019). The required sample amounts for our IR-MS system were over 6 nmol OCS. The overall precision value (1σ) for the atmospheric δ34S(OCS) value is 0.4 ‰. By contrast, the GC–MC–ICP-MS method (Said-Ahmad et al., 2017; Angert et al., 2019) has a similar precision of 0.6 ‰ but only requires 20 pmol of OCS. Consequently, the IR-MS method requires an OCS sample 300 times larger than that for the GC–MC–ICP-MS method. Therefore, our IR-MS method with a developed large-volume air sampling system has shortcomings for sample size and/or logistics for field campaigns. However, it is worth noting that benefits of our IR-MS method with its large-volume air sampling system include its potential application of multi-isotope measurements of OCS by measuring CO+ fragment ions for carbon and oxygen isotopes as well as S+ fragment ions.
For this study, we developed a new OCS sampling and purification system. OCS is extracted from 500 L of ambient air with a collection efficiency of almost over 95 % of OCS. The blank of the sampling and purification system was (0.30±0.16) nmol and memory effects were negligible. By comparison with the previously used syringe injection (Hattori et al., 2015) we demonstrated that any potential isotopic fractionation during sampling and purification is negligible. The analytical repeatability values (1σ) for the δ34S(OCS) value with more than 6 nmol for the commercial OCS samples and synthesized OCS samples were 0.2 ‰. We ascertained the δ34S(OCS) values for four atmospheric samples at the Suzukakedai campus of the Tokyo Institute of Technology located in Yokohama, Kanagawa, Japan. δ33S(OCS) values were not reported because of a small overlapping signal on m∕z 33 in the ambient air samples. The OCS concentrations and δ34S(OCS) values were in the range of 447–520 pmol mol−1 and 10.4 ‰–10.7 ‰, respectively. No clear diurnal variation in the δ34S(OCS) values was observed. Further modification of gas chromatographic techniques and/or data processing must be undertaken to measure δ33S(OCS) and Δ33S(OCS) values in future studies.
Earlier we proposed a δ34S(OCS) value of (4.9±0.3) ‰ for atmospheric OCS from measurements from a commercially available cylinder of compressed air (sample G in this study) (Hattori et al., 2015). Based on the four atmospheric samples taken in this study we revise this earlier value to (10.5±0.4) ‰, which is clearly distinct from the earlier value. The new δ34S(OCS) proposed here is in accordance with the δ34S(OCS) estimates of tropospheric and marine sources of OCS based on the OCS flux (Newman et al., 1991). Although OCS decomposition during preservation before the measurements was concerned, we found that no such OCS decomposition and isotopic fractionation have been observed for the modified adsorption tube with a Sulfinert-treated tube and valves and preservation at −80 ∘C within at least 90 days for OCS concentration and up to 14 days for δ34S(OCS) values.
Recently, Angert et al. (2019) reported the δ34S(OCS) value of ∼13 ‰ in Israel or the Canary Islands, and they suggested that the δ34S(OCS) value is homogeneous throughout the world. Although it is difficult to identify the reason for the difference of atmospheric δ34S(OCS) values between 10.5 ‰ in Japan and ∼13 ‰ in Israel or the Canary Islands, spatial variation and temporal variation of δ34S(OCS) values are expected to be a link between anthropogenic activities and OCS cycles.
The data presented in this paper is available in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/amt-12-1141-2019-supplement.
SH designed this research. KK and SH developed the system and performed the experiments. KK, SH, EB, and NY analysed the data. SH and KK contributed to the writing and revision of the paper with input from all co-authors.
The authors declare that they have no conflict of interest.
We thank Keita Yamada for his support of maintenance of the GC–Q-MS.
Thoughtful and constructive reviews by the three referees led to significant
improvements to the paper. This study was supported by JSPS KAKENHI (16H05884
(Shohei Hattori), 17J08979 (Kazuki Kamezaki), and 17H06105 (Naohiro Yoshida
and Shohei Hattori) from the Ministry of Education, Culture, Sports, Science
and Technology (MEXT), Japan. For system development, this study is supported
by research funds as a project formation support expenditure
“Internationalization of standards for quantification of biogeochemical
process with innovated isotopologue tracers” (Naohiro Yoshida) from the
Tokyo Institute of Technology. Enno Bahlmann acknowledges the Leibniz
Association SAW funding for the project “Marine biological production,
organic aerosol particles and marine clouds: a Process Chain (MarParCloud)”
(SAW-2016-TROPOS-2).
Edited by: Pierre Herckes
Reviewed by: Jan Kaiser and two anonymous referees
Amrani, A., Said-Ahmad, W., Shaked, Y., and Kiene, R. P.: Sulfur isotope homogeneity of oceanic DMSP and DMS, P. Natl. Acad. Sci. USA, 110, 18413–18418, 2013.
Angert, A., Said-Ahmad, W., Davidson, C., and Amrani A.: Sulfur isotopes ratio of atmospheric carbonyl sulfide constrains its sources, Sci. Rep., 9, 1–8, 2019.
Bahlmann, E., Weinberg, I., Seifert, R., Tubbesing, C., and Michaelis, W.: A high volume sampling system for isotope determination of volatile halocarbons and hydrocarbons, Atmos. Meas. Tech., 4, 2073–2086, https://doi.org/10.5194/amt-4-2073-2011, 2011.
Berkelhammer, M., Asaf, D. Still, C., Montzka, S., Noone, D., Gupta, M., Provencal, R., Chen, H., and Yakir, D.: Constraining surface carbon fluxes using in situ measurements of carbonyl sulfide and carbon dioxide, Global Biogeochem. Cy., 28, 161–179, https://doi.org/10.1002/2013GB004644, 2014.
Berry, J., Wolf, A., Campbell, J. E., Baker, I., Blake, N., Blake, D., Denning, A. S., Kawa, S. R., Montzka, S. A., Seibt, U., Stimler, K., Yakir, D., and Zhu, Z.: A coupled model of the global cycles of carbonyl sulfide and CO2: A possible new window on the carbon cycle, J. Geophys. Res.-Biogeosci., 118, 842–852, 2013.
Brenninkmeijer, C. A. M., Janssen, C., Kaiser, J., Rockmann, T., Rhee, T. S., and Assonov, S. S.: Isotope effects in the chemistry of atmospheric trace compounds, Chem. Rev., 103, 5125–5161, 2003.
Brühl, C., Lelieveld, J., Crutzen, P. J., and Tost, H.: The role of carbonyl sulphide as a source of stratospheric sulphate aerosol and its impact on climate, Atmos. Chem. Phys., 12, 1239–1253, https://doi.org/10.5194/acp-12-1239-2012, 2012.
Campbell, J. E., Carmichael, G. R., Chai, T., Mena-Carrasco, M., Tang, Y., Blake, D. R., Blake, N. J., Vay, S. A., Collatz, G. J., Baker, I., Berry, J. A., Montzka, S. A., Sweeney, C., Schnoor, J. L., and Stanier, C. O.: Photosynthetic Control of Atmospheric Carbonyl Sulfide During the Growing Season, Science, 322, 1085–1088, 2008.
Chin, M. and Davis, D. D.: A reanalysis of carbonyl sulfide as a source of stratospheric background sulfur aerosol, J. Geophys. Res., 100, 8993–9005, 1995.
Crutzen, P. J.: Possible importance of CSO for sulfate layer of stratosphere, Geophys. Res. Lett., 3, 73–76, 1976.
Ferm, R. J.: The Chemistry of Carbonyl Sulfide, Chem. Rev., 57, 621–640, 1957.
Glatthor, N., Höpfner, M., Baker, I. T., Berry, J., Campbell, J. E., Kawa, S. R., Krysztofiak, G., Leyser, A., Sinnhuber, B.-M., Stiller, G. P., Stonecipher, J., and von Clarmann, T.: Tropical sources and sinks of carbonyl sulfide observed from space, Geophys. Res. Lett., 42, 10082–10090, https://doi.org/10.1002/2015GL066293, 2015.
Hattori, S., Danielache, S. O., Johnson, M. S., Schmidt, J. A., Kjaergaard, H. G., Toyoda, S., Ueno, Y., and Yoshida, N.: Ultraviolet absorption cross sections of carbonyl sulfide isotopologues OC32S, OC33S, OC34S and O13CS: isotopic fractionation in photolysis and atmospheric implications, Atmos. Chem. Phys., 11, 10293–10303, https://doi.org/10.5194/acp-11-10293-2011, 2011.
Hattori, S., Schmidt, J. A., Mahler, D. W., Danielache, S. O., Johnson, M. S., and Yoshida, N.: Isotope Effect in the Carbonyl Sulfide Reaction with O(3P), J. Phys. Chem. A, 116, 3521–3526, 2012.
Hattori, S., Toyoda, A., Toyoda, S., Ishino, S., Ueno, Y., and Yoshida, N.: Determination of the Sulfur Isotope Ratio in Carbonyl Sulfide Using Gas Chromatography/Isotope Ratio Mass Spectrometry on Fragment Ions 32S+, 33S+, and 34S+, Anal. Chem., 87, 477–484, 2015.
Inomata, Y., Matsunaga, K., Murai, Y., Osada, K., and Iwasaka, Y. J.: Simultaneous measurement of volatile sulfur compounds using ascorbic acid for oxidant removal and gas chromatography – flame photometric detection, Chromatogr. A, 864, 111–119, 1999.
Johnson, M. S., Feilberg, K. L., von Hessberg, P., and Nielsen, O. J.: Isotopic processes in atmospheric chemistry, Chem. Soc. Rev., 31, 313–323, 2002.
Kamezaki, K., Hattori, S., Ogawa, T., Toyoda, S., Kato, H., Katayama, Y., and Yoshida, N.: Sulfur isotopic fractionation of carbonyl sulfide during degradation by soil bacteria, Environ. Sci. Technol., 50, 3537–3544, 2016.
Kato, H., Igarashi, Y., Dokiya, Y., and Katayama, Y.: Vertical distribution of carbonyl sulfide at Mt. Fuji, Japan, Water, Air, Soil Pollut., 223, 159–167, 2012.
Khan, M. A. H., Whelan, M. E., and Rhew R. C.: Analysis of low concentration reduced sulfur compounds (RSCs) in air: Storage issues and measurement by gas chromatography with sulfur chemiluminescence detection, Talanta, 88, 581–586, 2012.
Kremser, S., Jones, N. B., Palm, M., Lejeune, B., Wang, Y., Smale, D., and Deutscher, N. M.: Positive trends in Southern Hemisphere carbonyl sulfide, Geophys. Res. Lett., 42, 9473–9480, https://doi.org/10.1002/2015GL065879, 2015.
Kuai, L., Worden, J., Kulawik, S. S., Montzka, S. A., and Liu, J.: Characterization of Aura TES carbonyl sulfide retrievals over ocean, Atmos. Meas. Tech., 7, 163–172, https://doi.org/10.5194/amt-7-163-2014, 2014.
Lin, Y., Sim, M. S., and Ono, S.: Multiple-sulfur isotope effects during photolysis of carbonyl sulfide, Atmos. Chem. Phys., 11, 10283–10292, https://doi.org/10.5194/acp-11-10283-2011, 2011.
Montzka, S. A., Aydin, M., Battle, M., Butler, J. H., Saltzman, E. S., Hall, B. D., Clarke, A. D., Mondeel, D., and Elkins, J. W.: A 350-year atmospheric history for carbonyl sulfide inferred from Antarctic firn air and air trapped in ice, J. Geophys. Res., 109, D22302, https://doi.org/10.1029/2004JD004686, 2004.
Montzka, S. A., Calvert, P., Hall, B. D., Elkins, J. W., Conway, T. J., Tans, P. P., and Sweeney, C.: On the global distribution, seasonality, and budget of atmospheric carbonyl sulfide (COS) and some similarities to CO2, J. Geophys. Res.-Atmos., 112, D09302, https://doi.org/10.1029/2006JD007665, 2007.
Newman, L., Krouse, H. R., and Grinenko, V. A.: Stable Isotopes: NAACO; Scope; John Wiley and Sons, Chapter 5 Sulphur Isotope Variations Atmosphere, 133–176, 1991.
Oduro, H., Van Alstyne, K. L., and Farquhar, J.: Sulfur isotope variability of oceanic DMSP generation and its contributions to marine biogenic sulfur emissions, P. Natl. Acad. Sci. USA, 109, 9012–9016, 2012.
Ogawa, T., Hattori, S., Kamezaki, K., Kato, H., Yoshida, N., and Katayama, Y.: Isotopic fractionation of sulfur in carbonyl sulfide by carbonyl sulfide hydrolase of Thiobacillus thioparus THI115, Microbes Environ., 32, 367–375, 2017.
Ono, S., Shanks III, W. C., Rouxel, O. J., and Rumble, D.: S-33 constraints on the seawater sulfate contribution in modern seafloor hydrothermal vent sulfides, Geochim. Cosmochim. Ac., 71, 1170–1182, 2007.
Robinson, B. W.: Sulfur isotope standards, Reference and intercomparison materials for stable isotopes of light elements, in: Proceedings of a Consultants Meeting, Vienna, 1–3 December, 39–46, 1993.
Said-Ahmad, W. and Amrani, A.: A sensitive method for the sulfur isotope analysis of dimethyl sulfide and dimethylsulfoniopropionate in seawater, Rapid Commun. Mass Spectrom., 27, 2789–2796, 2013.
Said-Ahmad, W., Wong, K., Mcnall, M., Shawar, L., Jacksier, T., Turich, C., Stankiewicz, A., and Amrani, A.: Compound-Specific Sulfur Isotope Analysis of Petroleum Gases, Anal. Chem., 89, 3199–3207, 2017.
Sandoval-Soto, L., Stanimirov, M., von Hobe, M., Schmitt, V., Valdes, J., Wild, A., and Kesselmeier, J.: Global uptake of carbonyl sulfide (COS) by terrestrial vegetation: Estimates corrected by deposition velocities normalized to the uptake of carbon dioxide (CO2), Biogeosciences, 2, 125–132, https://doi.org/10.5194/bg-2-125-2005, 2005.
Schmidt, J. A., Johnson, M. S., Jung, Y., Danielache, S. O., Hattori, S., and Yoshida, N.: Predictions of the sulfur and carbon kinetic isotope effects in the OH + OCS reaction, Chem. Phys. Lett., 531, 64–69, 2012.
Schmidt, J. A., Johnson, M. S., Hattori, S., Yoshida, N., Nanbu, S., and Schinke, R.: OCS photolytic isotope effects from first principles: sulfur and carbon isotopes, temperature dependence and implications for the stratosphere, Atmos. Chem. Phys., 13, 1511–1520, https://doi.org/10.5194/acp-13-1511-2013, 2013.
Tcherkez, G. and Tea, I.: 32S∕34S isotope fractionation in plant sulphur metabolism, New Phytol., 200, 44–53, 2013.
Xu, X., Bingemer, H. G., and Schmidt, U.: The flux of carbonyl sulfide and carbon disulfide between the atmosphere and a spruce forest, Atmos. Chem. Phys., 2, 171–181, https://doi.org/10.5194/acp-2-171-2002, 2002.
Watts, S. F.: The mass budgets of carbonyl sulfide, dimethyl sulfide, carbon disulfide and hydrogen sulfide, Atmos. Environ., 34, 761–779, 2000.
Zuiderweg, A., Holzinger, R., Martinerie, P., Schneider, R., Kaiser, J., Witrant, E., Etheridge, D., Petrenko, V., Blunier, T., and Röckmann, T.: Extreme 13C depletion of CCl2F2 in firn air samples from NEEM, Greenland, Atmos. Chem. Phys., 13, 599–609, https://doi.org/10.5194/acp-13-599-2013, 2013.
Zumkehr, A., Hilton, T. W., Whelan, M., Smith, S., Kuai, L., Worden, J., and Campbell, J. E.: Global gridded anthropogenic emissions inventory of carbonyl sulfide, Atmos. Environ., 183, 11–19, 2018.