the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 17 Dec 2019
| 17 Dec 2019
Multi-scheme chemical ionization inlet (MION) for fast switching of reagent ion chemistry in atmospheric pressure chemical ionization mass spectrometry (CIMS) applications
Jyri Mikkilä
Siddharth Iyer
Jani Hakala
A novel chemical ionization inlet named the Multi-scheme chemical IONization inlet (MION), Karsa Ltd., Helsinki, Finland) capable of fast switching between multiple reagent ion schemes is presented, and its performance is demonstrated by measuring several known oxidation products from much-studied cyclohexene and α-pinene ozonolysis systems by applying consecutive bromide (Br−) and nitrate () chemical ionization. Experiments were performed in flow tube reactors under atmospheric pressure and room temperature (22 ∘C) utilizing an atmospheric pressure interface time-of-flight mass spectrometer (APi-ToF-MS, Tofwerk Ltd., Thun, Switzerland) as the detector. The application of complementary ion modes in probing the same steady-state reaction mixture enabled a far more complete picture of the detailed autoxidation process; the HO2 radical and the least-oxidized reaction products were retrieved with Br− ionization, whereas the highest-oxidized reaction products were detected in the mode, directly providing information on the first steps and on the ultimate endpoint of oxidation, respectively. While chemical ionization inlets with multiple reagent ion capabilities have been reported previously, an application in which the charging of the sample occurs at atmospheric pressure with practically no sample pretreatment, and with the potential to switch the reagent ion scheme within a second timescale, has not been introduced previously. Also, the ability of bromide ionization to detect highly oxygenated organic molecules (HOM) from atmospheric autoxidation reactions has not been demonstrated prior to this investigation.
- Article
(3098 KB) -
Supplement
(809 KB) - BibTeX
- EndNote





Chemical ionization mass spectrometry (CIMS) is a versatile analysis technique that enables detection of gas-phase molecular constituents at atmospheric pressure and at concentrations as low as 105 cm−3 (Munson and Field, 1966; Munson, 1977; Eisele and Tanner, 1993; Huey, 2007; de Gouw and Warneke, 2007; Mauldin et al., 2012; Sipilä et al., 2015; Hyttinen et al., 2018; Laskin et al., 2018). With the right selection of reagent ions that either form adducts with the analytes or transfer their electric charge (e.g., with an electron or a proton transfer), CIMS can offer a soft, selective, and extremely sensitive online detection for virtually any gas-phase chemical compounds. In recent years, various CIMS methods have revolutionized the ways we understand atmospheric volatile organic compound (VOC) oxidation processes (see e.g., Ehn et al., 2014; Rissanen et al., 2014; Jokinen et al., 2015; Kirkby et al., 2016; Lee et al., 2016; Breitenlechner et al., 2017), especially the routes leading to oxidative molecular growth, i.e., fast autoxidation (Crounse et al., 2013; Berndt et al., 2015; Rissanen et al., 2015) and slow aging (Donahue et al., 2006; Hallquist et al., 2009) and subsequent secondary organic aerosol (SOA) formation.
In principle, a mass spectrometer is a universal detector with applicability mainly controlled by two factors: (i) volatility, and (ii) ionizability of the analyte (Baeza-Romero et al., 2011; McLafferty, 2011). In practice, the sampled material must be volatilized into the gas phase and then ionized by a suitable method. In a gas-phase atmospheric application, the problem reduces to the latter as the analytes are inherently aloft in the surrounding gas media. Unfortunately, the most universal ionization methods, by definition, lack the selectivity needed for separating compounds from complex gas mixtures, and information from multiple complementary techniques are generally required to enable chemical speciation (e.g., isomer separation), with ambiguity nevertheless quickly increasing with the complexity of the target molecule. The utilization of multiple ionization schemes in CIMS has the potential for detailed chemical speciation of the target compounds by exploiting chemical selectivity, e.g., characterizing amines by ethanol CIMS (Yu and Lee, 2012), peroxy acids by (Iyer et al., 2017) and hydroperoxides by CF3O− (Crounse et al., 2006). However, CIMS instruments and their ion sources are expensive and bulky, and consequently most research laboratories have access to a precious few. Ideally, a versatile, easily deployable chemical ionization source capable of rapid switching between multiple reagent ions with varying chemical selectivity would be indispensable in tackling the complexity encountered in various gas-phase environments.
CIMS inlets with multiple reagent ion capabilities have been reported previously (e.g., Jordan et al., 2009; Brophy and Farmer, 2015), perhaps most commonly in applications concerning proton-transfer-reaction mass spectrometry (PTRMS). In addition to the usual H3O+ reagent ion (Hansel et al., 1995), NO+ and (Jordan et al., 2009), water clusters of H3O+ (i.e., (H2O)nH3O+) (Breitenlechner et al., 2017), (Zhang et al., 2019), and a few others (see (Blake et al., 2009) and references therein) have been employed. Selected ion flow tube (SIFT) applications have similarly utilized H3O+, NO+, and ionization schemes augmented further (in certain applications) by their fast switching and combined use of simultaneous reagent ions (Smith and Španel, 2005). For similar mass spectrometric detection as in the current work, Brophy and Farmer (Brophy and Farmer, 2015) have introduced a fast, switchable source for two concomitant ion mode operations and demonstrated it with the common low-pressure CIMS reagent ions acetate (CH3COO−) and iodide (I−). Also, other switchable ion sources have been introduced (e.g., Hearn and Smith, 2004; Agarwal et al., 2014; Pan et al., 2017) but with roughly similar utilization and performance characteristics as the ones presented above. In all these previously reported techniques the sample ionization occurs at reduced pressure, constituting sample pretreatment in drawing the atmospheric pressure material into the vacuum chambers of the mass spectrometer (MS) and consequently diluting the number concentration of the sample molecules considerably (i.e., by a factor of about 10 to 105). This renders almost any detection method useless in targeting analytes with very low gas-phase concentrations, such as ambient aerosol precursors sulfuric acid (SA) and individual highly oxidized multifunctional compounds (HOM, highly oxygenated organic molecules), which are present at around 105 to 107 cm−3 (Eisele and Tanner, 1993; Sipilä et al., 2010; Ehn et al., 2014; Rissanen et al., 2014; Jokinen et al., 2015; Kirkby et al., 2016). Also, dilution through a small orifice and subsequent turbulent mixing of ions into the sample in the low-pressure CIMS enhances recombination loss processes (e.g., radical–radical combination reactions and wall reactions of reactive and sticky compounds) causing depletion and subsequent detection bias. The current design is virtually devoid of these issues as the ionization occurs at atmospheric pressure by an ion insertion and the sample passes the ion source unperturbed.
While the utilization of multiple reagent ions within a single CIMS apparatus offers significant benefits, to this date it has not been reported in applications in which the ionization occurs at ambient pressure. Here we introduce a significant progress in the CIMS methodology by enabling atmospheric pressure sampling and ionization with multiple consecutive reagent ions in fast repetition, and without any pretreatment of the sampled gas mixture. When considering the detection of low-volatile, in situ aerosol precursor compounds such as HOM (i) that lack analytical standards, (ii) that have a range of individual detection sensitivities (Hyttinen et al., 2015, 2018), (iii) whose transmission and (iv) fragmentation are dependent on the detailed MS settings (Heinritzi et al., 2016; Zapadinsky et al., 2019; Passananti et al., 2019) and (v) whose detection is sensitive to changes in temperature (Frege et al., 2018), it becomes evident that retrieving complementary data depending on only one instrument calibration factor is extremely valuable. In the newly developed Multi-scheme chemical IONization inlet (MION) described here, the only changing parameter between the ionization stages is the ion-specific sensitivity to the target compounds as a function of the ion–molecule reaction time – which does not differ between applications. We report the characteristics and operation principle of this new inlet and compare its performance to previously reported CIMS inlets by coupling it to an atmospheric pressure time-of-flight mass spectrometer (APi-ToF-MS; Junninen et al., 2010) and measuring a multitude of previously reported oxidation products from much-studied cyclohexene and α-pinene ozonolysis systems (e.g., Yokouchi et al., 1985; Hatakeyama et al., 1985; Rissanen et al., 2014, 2015). To the best of our knowledge, the current type of an ambient pressure application in which the reagent ion scheme can be changed within a second timescale by simply switching a few voltages, has not been introduced previously.
A schematic of the Multi-scheme chemical IONization inlet, MION, (Karsa Ltd.) is presented in Fig. 1. The inlet consists of an electrically grounded 24 mm inner diameter (i.d.) flow tube with multiple coupled ion sources (simplified two-source setup shown in Fig. 1). The ion source connecting MION to the mass spectrometer entrance weighs about 2.1 kg with every additional stage weighing roughly 1.5 kg each. The distance of the first ion source to the mass spectrometer inlet orifice is approximately 3 cm, and the length of an additional ionization stage is about 10 cm, with a height of 16 cm. A schematic Fig. S1 with marked dimensions can be found in the Supplement. A gas-phase stream of nitrogen or air (both of purity > 99.99999 %, AGA) is enriched with the reagent ion precursor by feeding it through a saturator (or the precursor is obtained directly from a gas cylinder). The resulting reagent flows are then fed into their respective ion sources, where the reagents are ionized by a soft x-ray radiation (Hamamatsu L12535). The reagent ions are then accelerated and focused through 5 mm orifices into the laminar sample flow by electric fields. Small counterflows are applied through the orifices to prevent the mixing of the electrically neutral reagent precursors with the sample flow. Besides the ion source orifices, the flow tube design only minimally differs from tubular, making the flow pattern easy to define, the flow being essentially a flow through a circular pipe. The distance between the downstream ion source and the mass spectrometer pinhole is fixed, but the modular design of the MION allows the upstream source distance to be chosen to suit the application, with around 50 ms (milliseconds) minimum between ionization stages and longer times achievable by increasing the pipe length between the sources. For the experiments described in this paper the reaction time for the upstream reagent (nitrate ion, ) was 300 ms, and for the downstream reagent (bromide ion, Br−) 30 ms, when the total sample flow was 20 L min−1 (liters per minute).
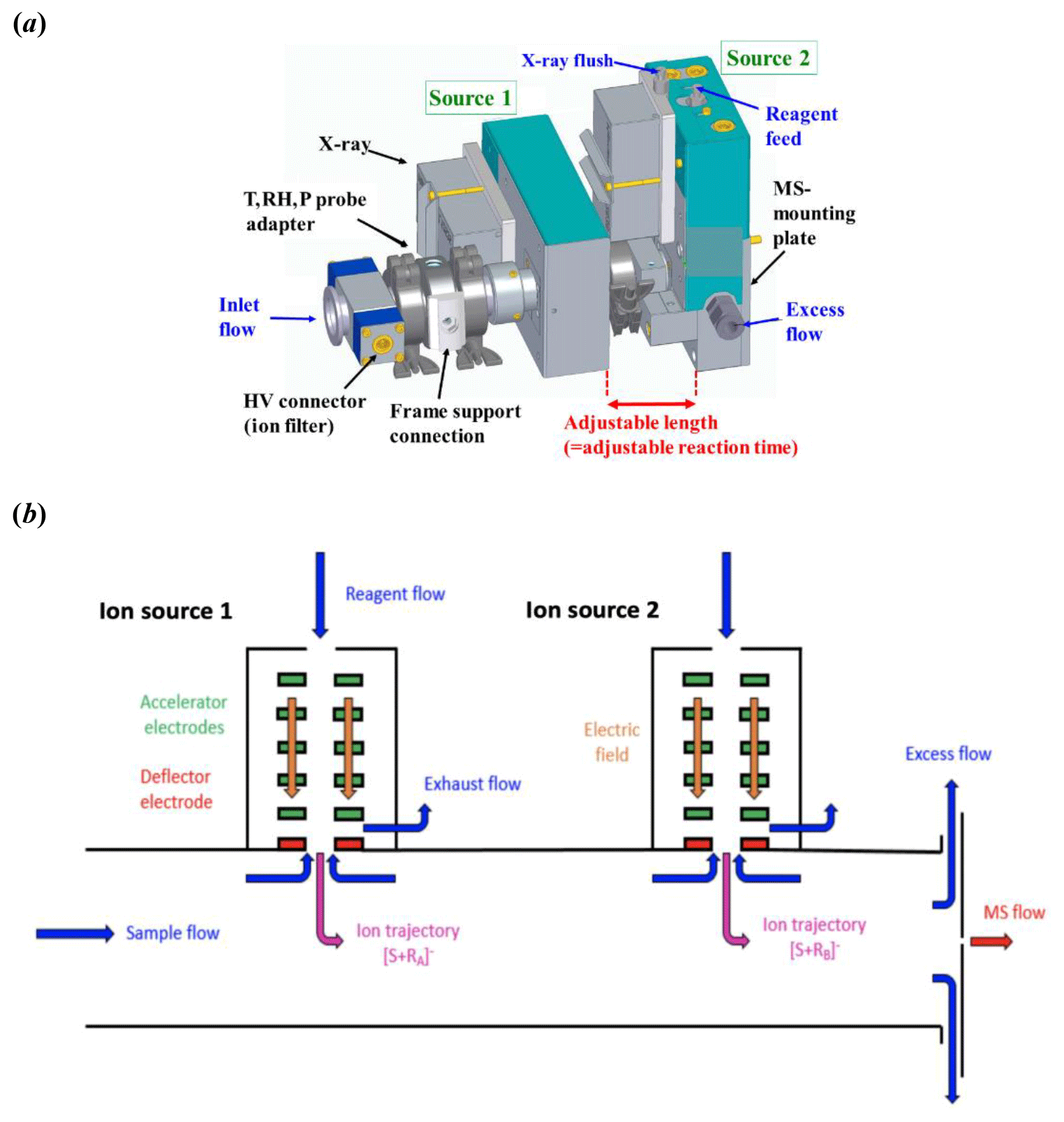
Figure 1The Multi-scheme chemical IONization inlet (MION). (a) Schematic description of the modular design shown with two concomitant ion sources with adjustable ion–molecule reaction time between them, coupled to an ion filter and meteorology sensor. (b) Illustration of the gas flows and ion paths within the inlet.
2.1 Operating the MION
The MION is operated by first turning on the counterflows for the ion sources, followed by turning on the sample flow, after which the reagent feed is turned on. The sample flow is controlled by a 50 L min−1 mass flow controller (mfc), and the counterflows and reagent feeds are controlled by 100 milliliters per minute (mL min−1) mfcs. The flow rates for the reagent feeds and counterflows depend on the compounds used as reagent ion precursors, but generally suitable counterflow (i.e., the counterflow setting subtracted by the reagent feed flow setting, producing a small suction to the sample flow) for the ion sources is as little as 25 mL min−1. After the flows have been chosen, the high voltage (ca. 2500 V) for the ion accelerator arrays is turned on. Lastly, to guide the reagent ions to the center of the sample flow, the ion deflector voltage (ca. 250 V) for the selected ion source is turned on. If the deflector electrode is grounded, it acts as an ion filter, and the ions produced in the ion source will not enter the sample flow. Selecting an ion mode (i.e., switching the ionization scheme) is as simple as turning on the deflector voltage for the desired ion source, i.e., turning on the deflector voltage in ion source A, while keeping the deflector in source B grounded, selects the ion chemistry of source A, and vice versa. Turning off (to 0 V) both deflectors will prevent all the ions from both of the ion sources from entering the sample flow, and thus the ambient ion mode is selected. Turning on both deflectors simultaneously will result in the downstream ion source ionization scheme to apply, as the downstream deflector will deflect most of the ions from the upstream one. While there may be settings where multiple chemical ionization schemes can work simultaneously, the application described in this paper was neither designed nor tested for that.
2.2 Switching the ion chemistry
The MION design enables rapid, concomitant application of multiple reagent ion chemistries in which selection and combination are mainly dictated by three variables: (i) target species to be ionized, (ii) characteristics of the reagent ion source compound, and (iii) the details of the ion–molecule reaction chemistry (e.g., adduct formation vs. charge transfer (Hyttinen et al., 2018)). By considering these issues it is possible to choose a combination of ionization schemes that critically supplement each other, significantly increasing the chemical information obtained from the targets. For example, measuring “a complete” product distribution with an inherently unselective reagent ion (e.g., bicarbonate, ) followed by a step-wise increase in chemical selectivity on further ionization stages (e.g., peroxy acids by (Iyer et al., 2017) and hydroperoxides by CF3O− (Crounse et al., 2006)). Similarly, adduct-forming reagent ions (e.g., ) can be augmented with ions strongly participating in further ion–molecule reactions with characteristic reaction and fragmentation patterns. An ion combination which works well with the same mass spectrometer settings between the ionization schemes is highly beneficial, as in this way the ion transmission characteristics of the instrument are minimally affected, and the detected signal heights depend mainly on the individual product ⋅ reagent ion binding strengths. This detection issue can be largely avoided if the two ionization schemes work with different ionization mechanisms, i.e., if the product detection sensitivity depends on the extent of fragmentation only in one mode and the other mode is barely influenced by the electric field strength change (e.g., if the first mode creates adducts and the second mode transfers charge, leading to ion adducts and charged molecules, respectively). Moreover, the design of the MION is ideally suited for investigating the detailed influence of ion–molecule reactions and reaction times (and thus also ion–molecule reaction kinetics) enabled by using the same reagent ion precursor feed for multiple ion sources. While in principle any combination of reagent ions is possible in the MION, for this work Br− and were selected not only due to their differing ionization characteristics and good performance under identical mass spectrometer settings, but also for their potential to offer complementary insight into the inspected VOC oxidation processes.
The fast switching between the ion modes is illustrated in Fig. 2, where the signals obtained for the reagent ions (i.e., Br− and ) and the total ion current (TIC) are shown for an experiment in which both ionization modes and a natural ion measurement (labeled APi in Fig. 2) were utilized. When neither source is active, the system measures only natural ambient ions, which results in a many orders of magnitude lower signal level. The rapid ion mode switch is completed within about a second time frame, with minimal interference from the idle ion mode. The small nitrate ion signal measured during bromide mode indicates that some nitric acid was present in the sampled gas stream and is ionized by Br−.
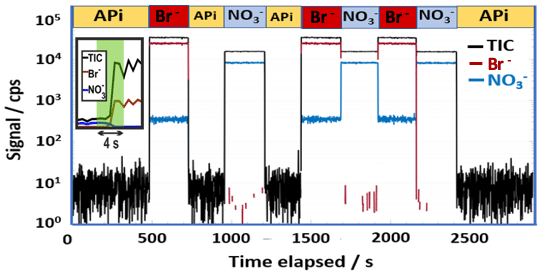
Figure 2An example of switching between multiple ion chemistries while sampling laboratory air. The ion modes utilized have been marked with separate colors and are further labeled above the figure with abbreviations: APi for ambient ion mode (no active ionization applied; black trace), Br− for bromide ion ionization mode (red trace), for nitrate ion ionization mode (blue trace), and TIC is used to indicate total ion count measured (black trace). The insert demonstrates the rapid ion mode switch, shown in a linear scale. All signals shown here were retrieved by high-resolution peak fitting.
Sensitivity of the MION was inspected by two independent ways: (i) by calibrating its response in both ion modes to photochemically produced sulfuric acid (SA, H2SO4) and (ii) by measuring oxidation products of ozonolysis-initiated autoxidation of cyclohexene and α-pinene, especially targeting HOMs (Rissanen et al., 2014, 2015). These very low-volatile, in situ atmospheric aerosol precursor compounds are typically present at around 105 to 107 cm−3 concentrations in the ambient gas phase, and thus an ability to directly detect them will testify the new inlet design's applicability for conducting field measurements. In addition, the SA calibration has been the standard method for estimating nitrate CIMS response to HOM detection (Ehn et al., 2014; Rissanen et al., 2015), which completely lacks any analytical standards, and the HOM product distributions of these two prototypical endocyclic alkenes are currently the best known.
3.1 Sulfuric acid calibration
Figure 3 shows the determined SA calibration plots, in which the measured H2SO4 is compared to values deduced by a reaction system simulation of OH-initiated SO2 photo-oxidation (see the Supplement for details). In both ion modes the MION detects gas-phase SA with good linearity of detection and detects well down to atmospheric concentration levels, with common sunlight daytime values generally ranging between 106 and 107 cm−3 (Eisele and Tanner, 1993; Sipilä et al., 2010). However, the sensitivities between the ion modes had about a factor of 10 difference, perhaps somewhat fortuitously, equaling the difference between the ion–molecule reaction times. The determined calibration factors were cm−3 for the nitrate mode with 300 ms ionization time and cm−3 for the bromide mode at 30 ms ionization, where the uncertainties refer to the statistical errors of the fits only. The overall uncertainty in the measured H2SO4 values obtained with this procedure was previously determined as 33 % (Kürten et al., 2012), and the resulting uncertainties in HOM detection have been reported previously as ±50 % (Ehn et al., 2014) or a factor of 2 (Berndt et al., 2015).
The obtained value compares well with the lower limit values reported previously for the nitrate CIMS employing APi-ToF-MS as the detector, which range roughly from 109 to 1010 cm−3 (e.g., Ehn et al., 2014; Rissanen et al., 2014; Berndt et al., 2015; Jokinen et al., 2015). For the Br− mode there are no previous values reported for SA detection. However, the HO2 radical detected with the bromide ionization well at roughly few parts per trillion (ppt) concentrations in the flow reactor experiments described next, is in accordance with the observations of Albrecht et al. (2019) and Sanchez et al. (2016), despite the differences in the ion source operating conditions (i.e., current ambient pressure ion source in contrast to the low-pressure sources used in the mentioned previous works).
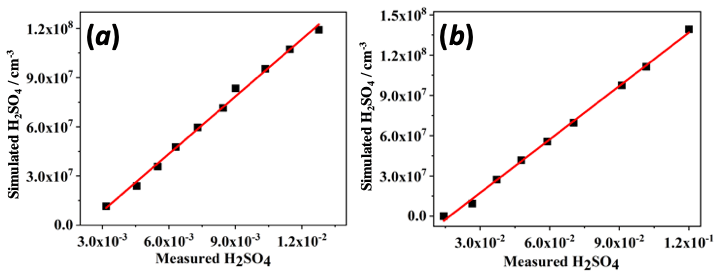
Figure 3Sulfuric acid (H2SO4) calibration plots showing the simulated values in molecule cm−3 against the measured, normalized ion signals determined for (a) Br− ion mode and for (b) ion mode. For the ion signal to concentration conversion see the Supplement.
3.2 VOC oxidation products
Cyclohexene (C6H10) and α-pinene (C10H16) ozonolysis-initiated autoxidation were investigated to inspect the applicability of the new inlet design to detect various reaction products with differing oxygen content, and to compare its performance to previously reported results obtained with other CIMS inlets (e.g., Ehn et al., 2014; Rissanen et al., 2014; Berndt et al., 2015; Mentel et al., 2015). The experiments were performed in quartz flow tube reactors (2.44 cm i.d. and 80 cm length, or 7.7 cm i.d. and 120 cm length) under atmospheric conditions with an inlet flow of 20 L min−1 for the MION, resulting in 2 to 10 s reaction times. The hydrocarbon precursor, at around 100 ppb in nitrogen (N2), was mixed with the bath gas air (N2+O2) and ozone (O3, ca. 50 ppb) a few centimeters upstream of the flow reactor. The introduction of ozone into the flow reactor containing the hydrocarbon precursor resulted in apparent instantaneous formation and subsequent detection of various HOM products, implying that rapid autoxidation of the endocyclic precursors took place, and that the new inlet design can detect these in situ aerosol precursors present at very low concentrations (i.e., fractions of ppt to several ppt for individual HOM products as reported previously for short reaction time conditions (Jokinen et al., 2015; Berndt et al., 2016)). The monomer and dimer HOM products detected here are covalently bound distinct molecules and do not result from ion–ion or ion–molecule reactions in the atmospheric pressure ionization inlet. In the recent literature, a considerable effort has been invested to unambiguously explain their origin and identity (see a recent review by Bianchi et al., 2019 and references therein). More details of the experimental setup and conditions can be found in the Supplement Sect. S2.
Typical spectra obtained with the MION during an α-pinene ozonolysis experiment in both ion modes are shown in Fig. 4 (for cyclohexene spectra see the Supplement); the bromide spectra have been shifted 17 mass units to illustrate common compositions on top of each other in the spectra, and the mass peak ranges have been divided into the reagent ion, monomer, and dimer mass ranges (see Figs. 4 and S4 in the Supplement). Inspecting the obtained spectra, it becomes immediately evident that only the bromide ionization retrieves few of the least-oxidized reaction products (but to a large extent also the further-oxidized compounds), whereas the extremely selective ionization is more sensitive to the most-oxidized species. However, in the present work, a 10 times longer ion–molecule reaction time was used for the mode, and thus the sensitivity to the higher-oxidized products is likely further augmented by this longer ionization time, which generally leads to increased sensitivity for the strongest bound reagent ⋅ product adducts, usually to the most-oxidized reaction products (Hyttinen et al., 2015, 2018). Naturally, the sensitivity changes as a function of the ion–molecule reaction time and that each ion–source distance needs to be calibrated to obtain their detailed response. In the MION setup, the ion–molecule reaction time is easily adjustable and thus is ideally suited to study these influences and even ion–molecule reaction kinetics. Nevertheless, the charging mechanism between these reagent ions differ; in nitrate ionization the analyte and HNO3 compete for , whereas in bromide ionization, Br− directly forms adducts with the analyte, i.e., the former is a ligand switching reaction while the latter is a direct adduct formation (Hyttinen et al., 2015, 2018), leading to inherently less selective ionization by Br−. Additionally, bromide ionization results in significantly more organic ion products devoid of the reagent ion, implying that other ion–molecule reactions are also involved in Br− ionization (e.g., deprotonation (Hansel et al., 2015; Breitenlechner et al., 2017) or dehydroxylation (Mielke et al., 2012; Iyer et al., 2017)) than simply adduct formation (see the Supplement for further details). In contrast, was only observed to deprotonate a few dicarboxylic acids and SA as reported in previous publications (Eisele et al., 1993; Rissanen et al., 2014). The organic ions were always minor in comparison to the most prominent product peaks.
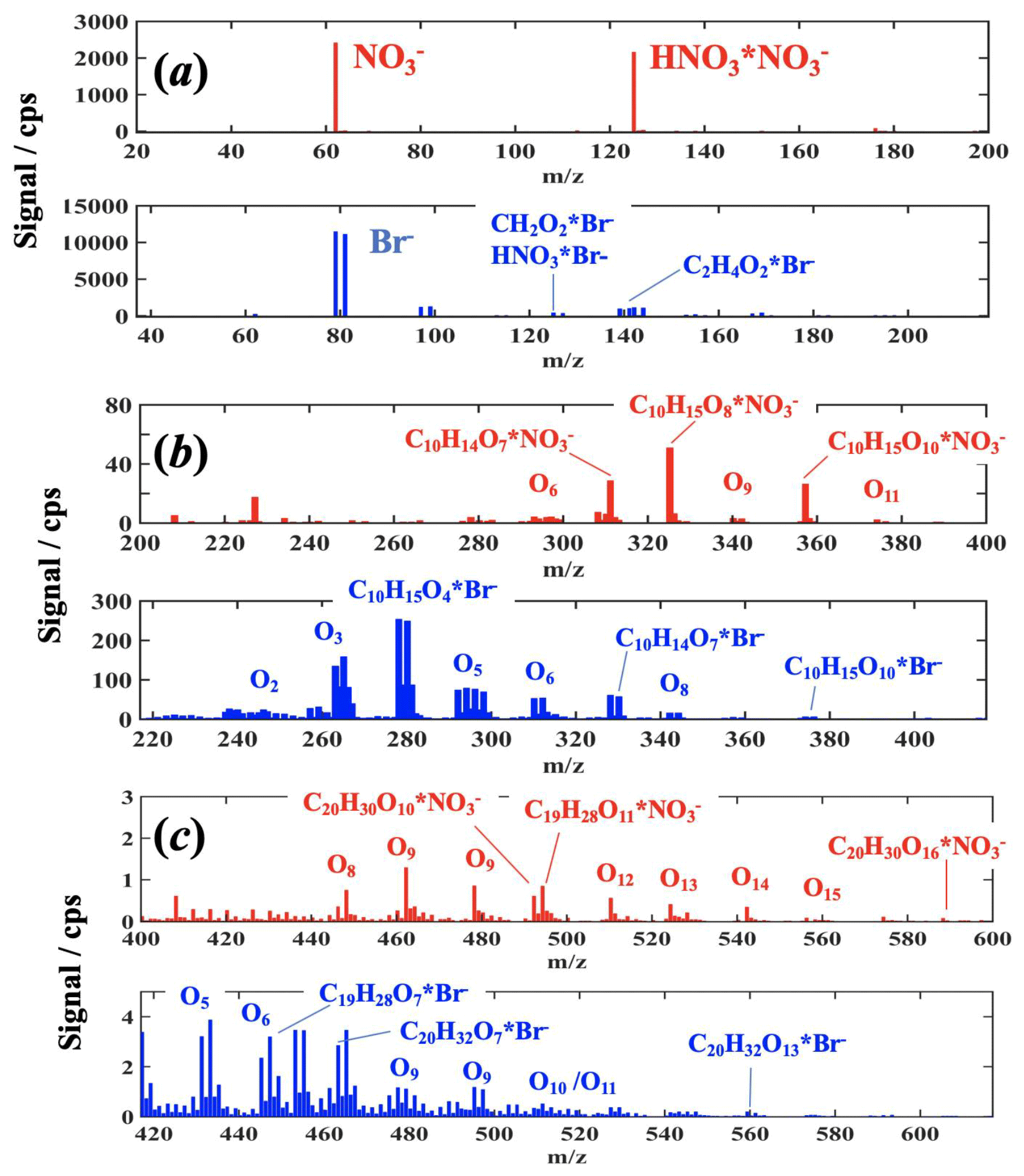
Figure 4Example MION spectra of organic oxidation products obtained from α-pinene ozonolysis experiments shown with a common product mass axis for both ion modes, i.e., the Br− spectrum (blue) is displaced by 17 Th (difference between reagent ion Br− and masses) to overlap the same composition products horizontally. (a) Illustrates the reagent ion spectra, (b) the monomer range (i.e., oxidation products which have the same number or less carbon atoms), and (c) the dimer range (i.e., oxidation products with about two times the carbon number of α-pinene), respectively. For a few most prominent products the explicit compositions are given, whereas for most of the detected peak groups only the average number of oxygen atoms is indicated; in dimer range the products with adjacent number of carbon and oxygen atoms start to overlap.
A critical aspect of using chemical ionization techniques is the sufficient availability of reagent ions. That is, when the analyte concentrations are too high, the observed reagent ion signal is considerably depleted. Under these conditions, the detection is rendered qualitative and it is not possible to determine the analyte concentrations. In the opposite situation, with too high primary ion production, there is a potential for secondary ion–ion reactions contributing to the measured apparent product ion signals. These finite limits exist for every chemical ionization inlet and are heavily dependent on the primary ion production rate and geometry of the ion source. For the current inlet system, we did not observe increased production of “dimer” or fragment products nor did we observe significant depletion of reagent ions, although a minor reduction in bromide reagent signal is evident when the highest concentration mixtures were sampled (at about 50 ppb of O3 and 500 ppb of α-pinene; see Figs. 5 and S2). However, this small reduction in bromide reagent ions did not deteriorate the linearity of the detection scheme (see Fig. S3). Furthermore, the measured product distributions closely resembled those that we have determined previously with an “Eisele–Tanner-type” inlet, verifying that if such influences were present they were minor at best.
The concomitant application of and Br− ionization is enabled by their comparable adduct binding strengths with oxidized organic molecules (Hyttinen et al., 2018). This means that both product-adducts are seen at similar efficiency, with the same mass spectrometer settings, and that strong adduct fragmentation would be seen in both ion modes if present. With very different product ⋅ adduct binding strengths, the least strongly bound adducts are likely lost to fragmentation in the APi section of the MS (Zapadinsky et al., 2019; Passananti et al., 2019). However, if the charging mechanism between the two reagent modes differ (e.g., one forms adducts and the other transfers charge), then they are likely applicable together and are largely unaffected by the mass spectrometer settings.
The fast switching of peroxy radical (RO2 and HO2) reagent ⋅ product adduct detection is illustrated in Fig. 5, where the ion signals obtained for the reagent ions and several α-pinene oxidation products are shown for a flow tube experiment conducted in air bath gas, at a 10 s residence time. High reactant concentrations were used, and the product signal levels are observed to change according to the VOC load of the flow reactor, marked in the figure by vertical dashed lines. The rapid ion mode change is completed within about 1-second time frame and is seen, for example, in the time traces measured for the prominent, highly oxidized peroxy radical formed by autoxidation, C10H17O17. This compound was detected well in both ion modes. Here, switching of the ion mode only shifts the mass of the product peak in the spectrum, with associated shift in signal intensity due to differences in the absolute detection sensitivities and total ion counts between the ion modes. In contrast, the HO2 radical and the primary ozonolysis derived peroxy radical, C10H15O4, are detected only in the Br− mode and thus disappear from the spectrum when nitrate mode is utilized. Similarly, few of the most-oxidized reaction products were prominent only in the spectra (Fig. 4). As seen in Fig. 5, some HNO3 was present in the sampled gas flow during the whole experiment, as the signal shows up during Br− stages. At the highest concentration experiments (at around 16:45), also small product signals were retrieved for the highly oxidized peroxy radicals attached to a nitrate reagent ion. Although this amount was enough to generate measurable product ion signals, it did not deteriorate the bromide ion detection (note that Br− will transfer its charge to very efficiently and leads to strong bromide ion signal depletion, which was not observed).
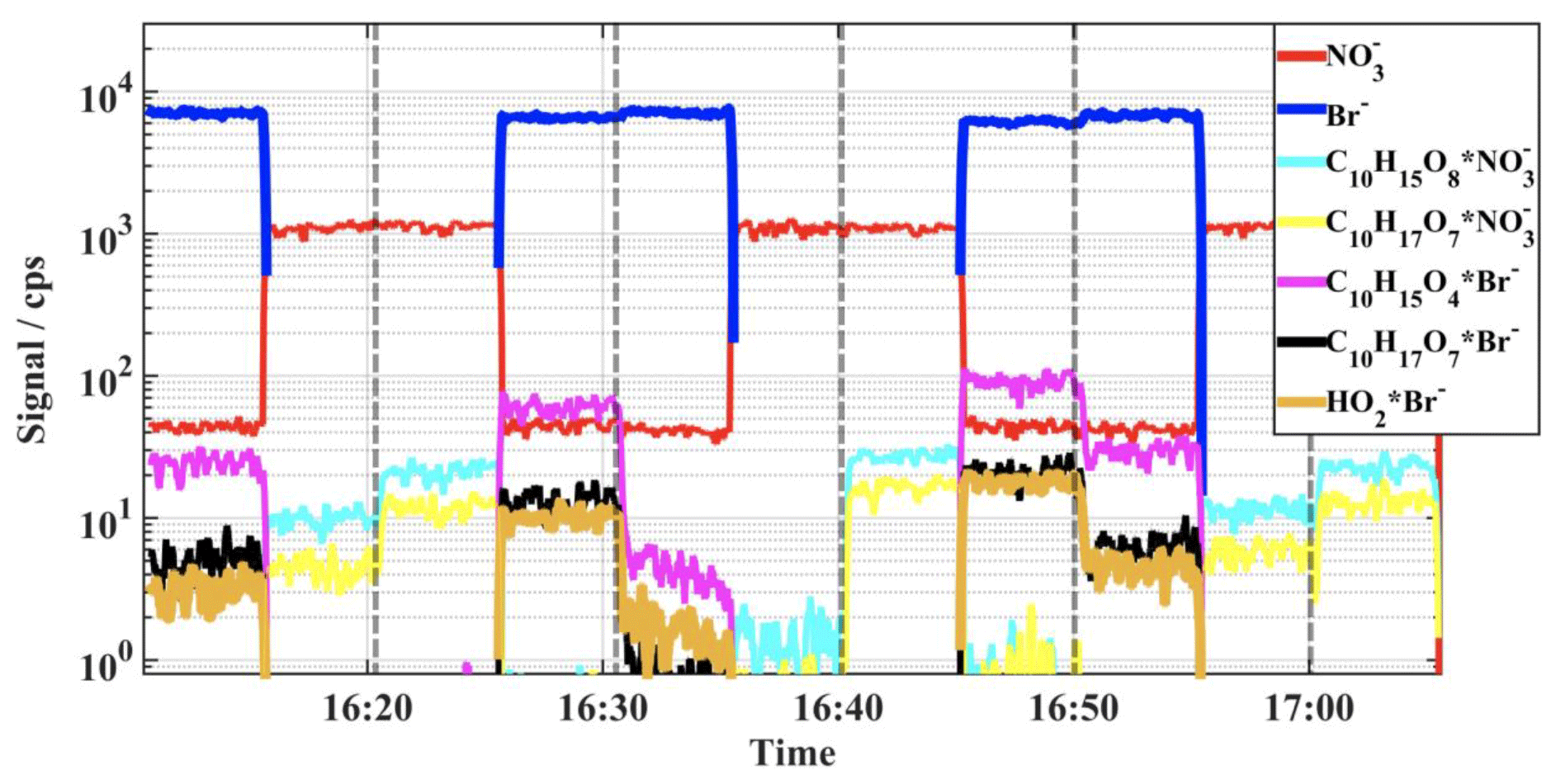
Figure 5Time traces measured for the reagent ions and selected α-pinene oxidation derived peroxy radicals illustrating the fast switching between reagent ion modes; the highly oxidized RO2 (C10H17O7) is retrieved well by both ion modes, whereas, the primary peroxy radicals HO2 and C10H15O4 were detected only with Br− ionization. The changes in the product signal levels are due to different hydrocarbon loads in the flow reactor, and the changes have been marked with vertical dashed lines. At the highest concentration experiments (at around 16:45; α-pinene about 500 ppb), small but measurable signals were retrieved also for the nitrate ion clustered products.
3.3 Comparison to previously reported cyclohexene and α-pinene HOM product distributions
The cyclohexene ozonolysis HOM product distribution reported previously in Rissanen et al. (2014), Berndt et al. (2015) and Mentel et al. (2015) is well recorded by the ion mode of the MION and even extends to the somewhat less-oxidized reaction products. This is likely due to the different ion injection process in the current inlet design which could potentially concomitantly decrease the sensitivity for the highest-oxidized compounds (Hyttinen et al., 2015). However, the observed sensitivity to the highest-oxidized species is similar within the measurement uncertainties to our previous works (e.g., Rissanen et al., 2014, 2015), and all the previously reported HOM by ionization are detected with the MION setup too.
In the bromide mode, also the least-oxidized reaction products down to two O-atoms were recorded, which amply illustrates the benefit of the multiple ion operation. To the best of our knowledge, this is the first time bromide ionization has been used to detect gas-phase cyclohexene and α-pinene ozonolysis products, and thus a direct comparison to previous results is not possible. Nevertheless, the Br− ionization retrieves even earlier reaction products in cyclohexene oxidation than those reported with the acetate (CH3COO−) (Berndt et al., 2015; Hyttinen et al., 2017) and iodide (I−) (Mielke et al., 2012; Iyer et al., 2017) reagent ions. For α-pinene, in addition to the HOMs reported previously by ionization (e.g., Ehn et al., 2014; Rissanen et al., 2015), bromide picks up several products with compositions matching the well-known, abundant, early generation oxidation products (e.g., pinic acid C9H14O4) (Ma et al., 2007) but which could also result from isomeric product compounds detected at the same exact mass. Additionally, ozonolysis of endocyclic alkenes produces a range of smaller carboxylic and peroxy acids as primary and secondary reaction products (Johnson and Marston, 2008), some of which were also detected in the current experiments with high sensitivity (e.g., based on the current alkene + O3 reaction rates, these products should be present at around maximum 1 ppb concentration with the long reaction time and high VOC loading experiments). Formic acid (HCOOH) and acetic acid (CH3COOH) were detected in the experiments, together with peaks having matching compositions to higher carboxylic acids, but which do not have unambiguous molecular compositions for definite mass specific identification. In addition, the strong acids, nitric acid (HNO3) and sulfuric acid (H2SO4), were also detected in the bromide mode, the latter used in calibration, and the former being a by-product of unrelated NOx experiments performed with the same inlet system.
3.4 Comparison to previously reported CIMS applications using multiple reagent ions
The MION inlet represents a significant improvement in measuring analytes at atmospheric pressure by employing multiple complementary ion chemistries. As mentioned above, to our knowledge, this is the only application in which the ionization occurs under ambient conditions and is completely devoid of any sample pretreatment. Perhaps closest to the current application is the two-ion-mode setup introduced by Brophy and Farmer (Brophy and Farmer, 2015), in which the ionization scheme is likewise changed by a simple ion optics voltage switching, but which operates under reduced pressures (roughly at 0.1 Atm), inherently lowering the sensitivity to the electrically neutral target compounds. In low-pressure inlets, the number concentration (i.e., the number of molecules per unit volume) of electrically neutral sample material is invariably diluted when pulled into the vacuum of the ionization chamber. While the low-pressure systems suffer from this dilution they generally generate significantly higher ion counts and, as such, do not experience as serious a reagent ion depletion as the ambient pressure sources. However, when targeting neutral analytes with very low ambient concentrations (e.g., HOMs and other in situ aerosol precursors) it is beneficial to avoid diluting the already low gas-phase concentration and, instead, apply atmospheric pressure ionization before the vacuum chambers. Crucially, the MION is not limited to the two-ion-mode operation described in the present paper and, due to its modular design, can accommodate as many reagent ions as feasible, only limited by space, utilizable ion–molecule reaction times, and other physicochemical constraints set by the reagent precursors.
The recently developed PTRMS instruments (Breitenlechner et al., 2017; Krechmer et al., 2018) can, in principle, rival the multi-ion operation due to their wide detection range for different oxidized states and their applicability for direct ambient pressure sampling. However, in practice, the product analysis is tedious due to the inherently low selectivity of the common H+ transfer reagents, resulting in a vast number of overlapping compounds. Yet again, the ionization in PTRMS instruments occurs at low pressures (commonly around 1 mbar), resulting in loss of sensitivity discussed above.
Atmospheric pressure chemical ionization mass spectrometry utilizing multiple reagent ion chemistries is a powerful analysis method for probing various gas-phase processes. The new type of inlet design introduced here enables selective inspection of gas-phase product distributions, allowed by rapid switching of chemical selectivity via specific reagent ion chemistries. This principle was demonstrated by detecting various previously reported oxidation products of cyclohexene and α-pinene applying Br− and ionization schemes in parallel. The sensitivity of the MION was observed to rival the previously reported CIMS inlets in detecting HOMs and sulfuric acid, exemplifying its applicability for field deployments. While the current setup was optimized for two-ion-mode operation, the modular construction of the MION enables it to, in principle, operate with multiple, unlimited ionization stages in fast repetition and without any instrument or sample pretreatment, the ion mode being changed by a simple switching of a few low-voltage settings.
Data will be available upon request from the corresponding authors.
The Supplement includes Sects. S1–S6: S1 Abstract – description of the supporting material, S2 Experimental setup and conditions, S3 Ion signal to concentration conversion, S4 Calibration measurement, S5 Product distributions – specific reaction products, S6 Organic ion peaks devoid of the reagent ion. Figures S1–S6; dimensions of the MION inlet and example spectra. The supplement related to this article is available online at: https://doi.org/10.5194/amt-12-6635-2019-supplement.
All authors contributed to designing and performing the experiments. Data analysis was done by MPR, SI and JM. MPR and JH wrote the paper. All authors discussed the results and commented on the paper.
Jyri Mikkilä and Jani Hakala work for Karsa, Ltd. Finland.
MPR is grateful for the support from the Academy of Finland (grant numbers 299574 and 326948).
This research has been supported by the Academy of Finland, Luonnontieteiden ja Tekniikan Tutkimuksen Toimikunta (grant nos. 299574 and 326948).
Open access funding provided by Helsinki University Library.
This paper was edited by Anna Novelli and reviewed by three anonymous referees.
Agarwal, B., Méndéz, R. G., Lanza, M., Sulzer, P., Märk, T. D., Thomas, N., and Mayhew, C. A.: Sensitivity and Selectivity of Switchable Reagent Ion Soft Chemical Ionization Mass Spectrometry for the Detection of Picric Acid, J. Phys. Chem. A, 118, 8229–8236, 2014.
Albrecht, S. R., Novelli, A., Hofzumahaus, A., Kang, S., Baker, Y., Mentel, T., Wahner, A., and Fuchs, H.: Measurements of hydroperoxy radicals (HO2) at atmospheric concentrations using bromide chemical ionisation mass spectrometry, Atmos. Meas. Tech., 12, 891–902, https://doi.org/10.5194/amt-12-891-2019, 2019.
Baeza-Romero, M. T., Blitz, M. A., Goddard, A., and Seakins, P. W.: Time-of-flight mass spectrometry for time-resolved measurements: Some developments and applications, Int J. Chem. Kin., 44, 532–545, 2011.
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-Phase Ozonolysis of Cycloalkenes: Formation of Highly Oxidized RO2 Radicals and Their Reactions with NO, NO2, SO2, and Other RO2 Radicals, J. Phys. Chem. A, 119, 10336–10348, 2015.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, 2019.
Blake, R. S., Monks, P. S., and Ellis, A. M.: Proton-Transfer Reaction Mass Spectrometry, Chem. Rev., 109, 861–896, 2009.
Breitenlechner, M., Fischer, L., Hainer, M., Heinritzi, M., Curtius, J., and Hansel, A.: PTR3: An Instrument for Studying the Lifecycle of Reactive Organic Carbon in the Atmosphere, Anal. Chem., 89, 5824–5831, 2017.
Brophy, P. and Farmer, D. K.: A switchable reagent ion high resolution time-of-flight chemical ionization mass spectrometer for real-time measurement of gas phase oxidized species: characterization from the 2013 southern oxidant and aerosol study, Atmos. Meas. Tech., 8, 2945–2959, https://doi.org/10.5194/amt-8-2945-2015, 2015.
Crounse, J. D., McKinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of Gas-Phase Hydroperoxides by Chemical Ionization Mass Spectrometry, Anal. Chem., 78, 6726–6732, 2006.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, 2013.
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry, Mass. Spectrom. Rev., 26, 223-257, 2007.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled Partitioning, Dilution, and Chemical Aging of Semivolatile Organics, Environ. Sci. Technol., 40, 2635–2643, 2006.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, 2014.
Eisele, F. L. and Tanner, D. J.: Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res.-Atmos., 98, 9001–9010, 1993.
Frege, C., Ortega, I. K., Rissanen, M. P., Praplan, A. P., Steiner, G., Heinritzi, M., Ahonen, L., Amorim, A., Bernhammer, A.-K., Bianchi, F., Brilke, S., Breitenlechner, M., Dada, L., Dias, A., Duplissy, J., Ehrhart, S., El-Haddad, I., Fischer, L., Fuchs, C., Garmash, O., Gonin, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kirkby, J., Kürten, A., Lehtipalo, K., Leiminger, M., Mauldin, R. L., Molteni, U., Nichman, L., Petäjä, T., Sarnela, N., Schobesberger, S., Simon, M., Sipilä, M., Stolzenburg, D., Tomé, A., Vogel, A. L., Wagner, A. C., Wagner, R., Xiao, M., Yan, C., Ye, P., Curtius, J., Donahue, N. M., Flagan, R. C., Kulmala, M., Worsnop, D. R., Winkler, P. M., Dommen, J., and Baltensperger, U.: Influence of temperature on the molecular composition of ions and charged clusters during pure biogenic nucleation, Atmos. Chem. Phys., 18, 65–79, https://doi.org/10.5194/acp-18-65-2018, 2018.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hansel, A., Jordan, A., Holzinger, R., Prazeller, P., Vogel, W., and Lindinger, W.: Proton transfer reaction mass spectrometry: on-line trace gas analysis at the ppb level, Int. J. Mass Spectrom. Ion Process., 149, 609–619, 1995.
Hatakeyama, S., Tanonaka, T., Weng, J.-H., Bandow, H., Takagi, H., and Akimoto, H.: Ozone-Cyclohexene Reaction in Air: Quantitative Analysis of Particulate Products and the Reaction Mechanism, Environ. Sci. Technol., 19, 935–942, 1985.
Hearn, J. D. and Smith, G. D.: A Chemical Ionization Mass Spectrometry Method for the Online Analysis of Organic Aerosols, Anal. Chem., 76, 2820–2826, 2004.
Heinritzi, M., Simon, M., Steiner, G., Wagner, A. C., Kürten, A., Hansel, A., and Curtius, J.: Characterization of the mass-dependent transmission efficiency of a CIMS, Atmos. Meas. Tech., 9, 1449–1460, https://doi.org/10.5194/amt-9-1449-2016, 2016.
Huey, L. G.: Measurement of trace atmospheric species by chemical ionization mass spectrometry: Speciation of reactive nitrogen and future directions, Mass Spectrom. Rev., 26, 166–184, 2007.
Hyttinen, N., Kupiainen-Määttä, O., Rissanen, M. P., Muuronen, M., Ehn, M., and Kurtén, T.: Modeling the Detection of Highly Oxidized Cyclohexene Ozonolysis Products Using Nitrate-Based Chemical Ionization, J. Phys. Chem. A, 119, 6339–6345, 2015.
Hyttinen, N., Rissanen, M. P., and Kurtén, T.: Computational Comparison of Acetate and Nitrate Chemical Ionization of Highly Oxidized Cyclohexene Ozonolysis Intermediates and Products, J. Phys. Chem. A, 121, 2172–2179, 2017.
Hyttinen, N., Otkjær, R. V., Iyer, S., Kjaergaard, H. G., Rissanen, M. P., Wennberg, P. O., and Kurtenì, T.: Computational Comparison of Different Reagent Ions in the Chemical Ionization of Oxidized Multifunctional Compounds, J. Phys. Chem. A, 122, 269–279, 2018.
Iyer, S., He, X., Hyttinen, N., Kurtén, T., and Rissanen, M. P.: Computational and Experimental Investigation of the Detection of HO2 Radical and the Products of Its Reaction with Cyclohexene Ozonolysis Derived RO2 Radicals by an Iodide-Based Chemical Ionization Mass Spectrometer, J. Phys. Chem. A, 121, 6778–6789, 2017.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, 2008.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, 2015.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Herbig, J., Märk, L., Schottkowsky, R., Seehauser, H., Sulzer, P., and Märk, T. D.: An online ultra-high sensitivity Proton-transfer-reaction mass-spectrometer combined with switchable reagent ion capability (PTR + SRI − MS), Int. J. Mass Spectrom., 286, 32–38, 2009.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junnxinen, H., Kangasluoma, J., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Peräkylä, O., Piel, F., Petäjä, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipilä, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tomé, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, 2016.
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and de Gouw, J.: Evaluation of a New Reagent-Ion Source and Focusing Ion–Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, 2018.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Calibration of a Chemical Ionization Mass Spectrometer for the Measurement of Gaseous Sulfuric Acid, J. Phys. Chem. A, 116, 6375–6386, 2012.
Laskin, J., Laskin, A., and Nizkorodov, S. A.: Mass Spectrometry Analysis in Atmospheric Chemistry, Anal. Chem., 90, 166–189, 2018.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Joste, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H. Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, 2016.
Ma, Y., Willcox, T. R.; Russell, A. T., and Marston, G.: Pinic and pinonic acid formation in the reaction of ozone with a-pinene, Chem. Comm., 13, 1328–1330, 2007.
Mauldin III, R. L., Berndt, T., Sipilä, M., Paasonen, P., Petäjä, T., Kim, S., Kurtén, T., Stratmann, F., Kerminen, V.-M., and Kulmala, M.: A new atmospherically relevant oxidant of sulphur dioxide, Nature, 488, 193–196, 2012.
McLafferty, F. D.: A Century of Progress in Molecular Mass Spectrometry, Annu. Rev. Anal. Chem., 4, 1–22, 2011.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Mielke, L. H. and Osthoff, H. D.: On quantitative measurements of peroxycarboxylic nitric anhydride mixing ratios by thermal dissociation chemical ionization mass spectrometry, Int. J. Mass Spectrom., 310, 1–9, 2012.
Munson, B.: Chemical Ionization Mass Spectrometry, Anal. Chem., 49, 772A–778A, 1977.
Munson, M. S. B. and Field, F. H.: Chemical Ionization Mass Spectrometry I: General Introduction, J. Am. Chem. Soc., 88, 2621–2630, 1966.
Pan, Y., Zhang, Q., Zhou, W., Zou, X., Wang, H., Huang, C., Shen, C., and Chu, Y.: Detection of Ketones by a Novel Technology: Dipolar Proton Transfer Reaction Mass Spectrometry (DP-PTR-MS), J. Am. Soc. Mass Spectrom., 28, 873–879, 2017.
Passananti, M., Zapadinsky, E., Zanca, T., Kangasluoma, J., Myllys, N., Rissanen, M P., Kurtén, T., Ehn, M., Attoui, M., and Vehkamäki, H.: How well can we predict cluster fragmentation inside a mass spectrometer?, Chem. Commun., 55, 5946–5949, 2019.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, 2014.
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kausiala, O., Garmash, O., Kjaergaard, H. G., Petä jä, T., Worsnop, D. R., Ehn, M., and Kulmala, M.: Effects of Chemical Complexity on the Autoxidation Mechanisms of Endocyclic Alkene Ozonolysis Products: From Methylcyclohexenes toward Understanding α-Pinene, J. Phys. Chem. A, 119, 4633–4650, 2015.
Sanchez, J., Tanner, D. J., Chen, D., Huey, L. G., and Ng, N. L.: A new technique for the direct detection of HO2 radicals using bromide chemical ionization mass spectrometry (Br-CIMS): initial characterization, Atmos. Meas. Tech., 9, 3851–3861, https://doi.org/10.5194/amt-9-3851-2016, 2016.
Sipilä, M., Sarnela, N., Jokinen, T., Junninen, H., Hakala, J., Rissanen, M. P., Praplan, A., Simon, M., Kürten, A., Bianchi, F., Dommen, J., Curtius, J., Petäjä, T., and Worsnop, D. R.: Bisulfate – cluster based atmospheric pressure chemical ionization mass spectrometer for high-sensitivity (<100 ppqV) detection of atmospheric dimethyl amine: proof-of-concept and first ambient data from boreal forest, Atmos. Meas. Tech., 8, 4001–4011, https://doi.org/10.5194/amt-8-4001-2015, 2015.
Sipilä, M., Berndt, T., Petäjä, T., Brus, D., Vanhanen, J., Stratmann, F., Patokoski, J., and Mauldin III, R. L., Hyvärinen, A. P., Lihavainen, H., and Kulmala, M.: The role of sulfuric acid in atmospheric nucleation, Science, 327, 1243–1246, 2010.
Smith, D. and Španel, P.: Selected ion ?ow tube mass spectrometry (SIFT-MS) for on-line trace gas analysis, Mass Spectrom. Rev., 24, 661–700, 2005.
Yokouchi, Y. and Ambe Y.: Aerosols formed from the chemical reaction of monoterpenes and ozone, Atmos. Environ., 19, 1271–1276, 1985.
Yu, H. and Lee, S-H.: Chemical ionisation mass spectrometry for the measurement of atmospheric amines, Environ. Chem., 9, 190–201, 2012.
Zapadinsky, E., Passananti, M., Myllys, N., Kurtén, T., and Vehkamäki H.: Modeling on Fragmentation of Clusters inside a Mass Spectrometer, J. Phys. Chem. A., 123, 611–624, 2019.
Zhang, Q., Zou, X., Liang, Q., Wang, H., Huang, C., Shen, C., and Chu, Y.: Ammonia-Assisted Proton Transfer Reaction Mass Spectrometry for Detecting Triacetone Triperoxide (TATP) Explosive, J. Am. Soc. Mass Spectrom., 30, 501–508, 2019.